
Abstract
Introduction:
Ataxia-telangiectasia (AT) is an autosomal recessive neurodegenerative disease characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, humoral and cellular immunodeficiency, sensitivity to ionizing radiation, and a tendency to malignancy. The aim of this study was to evaluate the demographic characteristics, immunodeficiency status, laboratory findings, and prognosis of children with AT based on a single-center experience.
Patients and Methods:
Nineteen pediatric patients diagnosed with AT between 2003 and 2024 were retrospectively analyzed at a single tertiary center.
Results:
The study included 11 male and 8 female patients, with a mean follow-up duration of 72.6 ± 41.3 months. The mean ages at symptom onset and diagnosis were 30.47 ± 23.7 months and 82.32 ± 26.5 months, respectively. The most common presenting symptoms were gait instability, ocular telangiectasia, and recurrent respiratory tract infections. Immunological evaluation revealed IgA deficiency in 57.9% of the patients and IgG deficiency in 15.8%. A hyper-IgM AT phenotype was identified in two patients (10.5%). During follow-up, bronchiectasis and hepatosteatosis developed in 26.3% of the patients. One patient (5.2%) developed type 2 diabetes mellitus, and malignancy occurred in three patients (15.7%). Overall, 6 patients (31.5%) died during the follow-up period.
Conclusion:
Ataxia-telangiectasia is a rare multisystem disorder associated with significant morbidity and mortality. Early diagnosis, comprehensive multidisciplinary follow-up, regular malignancy surveillance, and preventive strategies for recurrent sinopulmonary infections are essential for improving clinical outcomes and prognosis in affected patients.
Introduction
Ataxia-telangiectasia (AT) is a rare autosomal recessive multisystem disorder characterized by progressive neurodegeneration, immunodeficiency, hypersensitivity to ionizing radiation, and a markedly increased risk of malignancy. The incidence of AT ranges from 1 in 40,000 to 1 in 300,000 live births worldwide.1,2 Ataxia-telangiectasia results from pathogenic variants in the ataxia-telangiectasia mutated (ATM) gene, which encodes a serine/threonine kinase essential for DNA repair and maintenance of genomic stability. 3
Progressive cerebellar ataxia is typically the earliest manifestation and becomes evident in early childhood, followed by worsening motor impairment and loss of ambulation in later years. Telangiectasia, most commonly involving the conjunctivae and sun-exposed skin, represents another characteristic feature. However, beyond these hallmark findings, AT is a clinically heterogeneous condition with significant multisystem involvement.4,5 Various additional dermatologic findings, such as café-au-lait macules and other pigmentary changes, have also been described. 6
Inborn errors of immunity are present in approximately 60%–80% of patients and represent a major cause of morbidity and mortality.7,8 Both humoral and cellular immune defects may occur, including lymphopenia and impaired antibody production, predisposing patients to recurrent and often severe sinopulmonary infections beginning early in life.9,10 These infections may lead to chronic pulmonary complications such as bronchiectasis and interstitial lung disease, which significantly influence long-term outcomes.7,11
Beyond neurological and immunological involvement, AT is associated with endocrine and metabolic abnormalities, including growth failure, hepatic steatosis, and glucose metabolism disorders, particularly during adolescence and adulthood.12,13
Due to defective DNA repair and increased radiosensitivity, patients with AT have a markedly elevated risk of malignancy. Hematological cancers predominate in childhood, whereas solid tumors are more frequent later in life.14,15 Recent cohort studies have confirmed the persistent cancer risk throughout the lifespan. 16
Management of AT remains largely supportive and multidisciplinary. Preventive strategies focus on minimizing infectious complications through vaccination, prophylactic antibiotic therapy, and intravenous immunoglobulin (IVIG) replacement in patients with significant antibody deficiency or recurrent infections. 8
Despite growing knowledge of the molecular basis of AT, its clinical course remains highly variable. Well-characterized single-center cohorts remain essential for understanding disease heterogeneity and long-term prognosis. The present study aimed to evaluate the demographic characteristics, immunological profile, multisystem manifestations, and prognosis of children with AT followed at a single center.
Patients and Methods
The medical records of patients with AT diagnosed between 2003 and 2024 at a single center were retrospectively analyzed. All patients were diagnosed according to the criteria of the European Society for Immunodeficiencies. This retrospective study was conducted at a tertiary pediatric referral center. Ethical approval was obtained from the local Institutional Review Board. Written informed consent was obtained from the parents or legal guardians of all participants in accordance with institutional and national ethical standards.
Patients’ gender and age at symptom onset and diagnosis, duration of follow-up, parental consanguinity and family history, diagnostic findings at presentation, physical examination findings, laboratory findings, results of genetic analyses, complications during clinical follow-up, and treatment regimens were evaluated. Complete blood count, alpha-fetoprotein levels, immunoglobulin levels, and lymphocyte subsets were assessed at diagnosis and during follow-up. Low immunoglobulin levels were defined as values of more than 2 standard deviations (SD) below the age-adjusted mean. 17 Lymphocyte immunophenotyping was performed using a flow cytometer, and lymphocyte subset percentages and absolute counts below the 5th percentile for age were considered low. 18 Serum AFP levels greater than 8 µg/L were considered significantly elevated in children aged 2 years and older. Descriptive statistics were used to summarize the data. Continuous variables were expressed as mean ± standard deviation or median (minimum–maximum), as appropriate, while categorical variables were presented as numbers and percentages. Statistical analyses were performed using SPSS (Statistical Package for Social Sciences; IBM Corp., Chicago, Illinois, USA), version 26.
Results
Nineteen patients with AT were included in the study, of whom 57.9% were male. The mean age at symptom onset was 30.47 ± 23.7 months, the mean age at diagnosis was 82.32 ± 26.5 months, and the mean follow-up duration was 72.6 ± 41.3 months. Parental first-degree consanguinity was present in 68.4% of the patients. A positive family history of AT was identified in 26.3% of the patients. The demographic characteristics are summarized in Table 1.
Demographic Characteristics of Patients with Ataxia-Telangiectasia

The most frequent presenting complaints were gait instability (73.6%), neuromotor delay (63.1%), and recurrent upper and lower respiratory tract infections (63.1%). Upon physical examination, all patients exhibited neurological ataxia and ocular telangiectasia. Growth parameters were below 2 SD for age and sex in 78.9% of the patients. Café-au-lait macules were observed in 31.5% of the patients. Additional findings, including cutaneous herpes simplex infection, finger clubbing, and a cushingoid appearance secondary to corticosteroid therapy, were each observed in a single, different patient.
At diagnosis, the median hemoglobin level was 11.8 g/dL, the median absolute lymphocyte count was 2.28 × 10³/µL, and the median absolute neutrophil count was 5.60 × 10³/µL. Serum AFP levels were elevated in all patients. A genetic analysis of the ATM gene was performed in 13 patients (68.4%). Homozygous mutations were identified in 12 patients, and a compound heterozygous mutation was identified in 1 patient.
Immunological evaluation demonstrated humoral and cellular immune abnormalities. IgA deficiency was detected in 57.9% of the patients, while IgG levels were below −2 SD in 15.8% of them. Elevated IgM levels were observed in 10.5% of patients. IgE levels were within normal limits in all patients at diagnosis. IgG subclass analysis revealed IgG2 deficiency in 75.0% of the evaluated patients. During follow-up, 21.1% of patients with initially normal IgG levels subsequently developed hypogammaglobulinemia.
Antibodies to hepatitis B surface antigen (anti-HBs) levels were measured in 13 of the 19 patients. Anti-tetanus antibody levels were measured in 9 patients, of whom 7 (77.8%) had an inadequate vaccine response. Three patients demonstrated inadequate responses to both vaccines.
Flow cytometric analysis revealed reduced CD19+ B-cell counts in 63.2% of the patients, reduced CD3+ T-cell counts in 21.1%, reduced CD4+ T-cell counts in 26.3%, and reduced CD8+ T-cell counts in 5.3%. At diagnosis, 3 patients (15.8%) had combined T- and B-lymphocyte counts below the 5th percentile for age. During follow-up, lymphocyte subset abnormalities developed in an additional 15.8% of the patients. The lymphocyte subset findings are summarized in Table 2.
Immunological Findings of Patients with Ataxia-Telangiectasia at Initial Evaluation
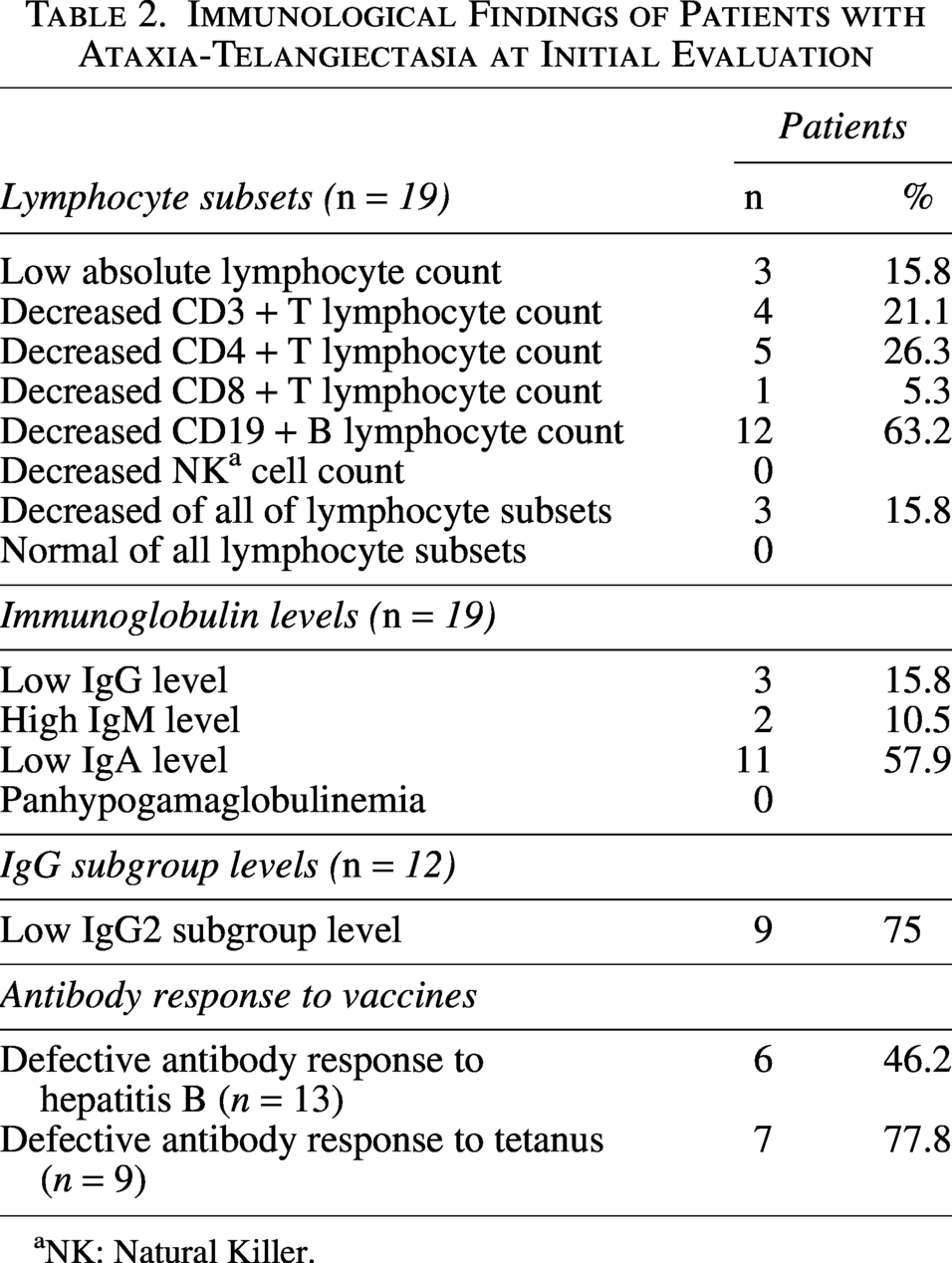
NK: Natural Killer.
Neuroimaging data were available for 12 of the 19 patients included in the study. Cerebellar atrophy consistent with AT was observed in 10 of these patients (83.3%). Neuroepithelial cystic formation in the left temporal lobe was observed in one patient, and mega cisterna magna was detected in another. Thoracic computed tomography was performed in 7 patients. Bronchiectasis was detected in 5 of these patients (71.4%). Three patients had bronchiectasis at the time of diagnosis, while 2 additional patients developed bronchiectasis during follow-up, during the first and eighth years of follow-up, respectively. Among the patients with bronchiectasis, the identified pathogens included Aspergillus fumigatus (n = 1), Mycobacterium tuberculosis (n = 2), and Pseudomonas aeruginosa (n = 2).
One patient had hepatosteatosis at the time of diagnosis, and four additional patients developed this condition during follow-up, resulting in an overall prevalence of 26.3%. Among these 5 patients, 3 were male and 2 female. The median age at detection of hepatosteatosis was 12 years (range, 7–17 years). One patient with hepatosteatosis subsequently developed type 2 diabetes mellitus during the follow-up period. The diagnosis of hepatosteatosis in all patients was established by abdominal ultrasonography, demonstrating increased hepatic echogenicity consistent with fatty infiltration. No patients underwent liver biopsy.
As an autoimmune manifestation, immune cytopenia was observed in 1 patient prior to the diagnosis of AT. This patient had been followed since infancy due to autoimmune hemolytic anemia and immune thrombocytopenia and later underwent immunological evaluation due to recurrent respiratory tract infections, growth retardation, and gait imbalance. Immunological assessment revealed markedly decreased serum IgG and IgA levels with elevated IgM (579 mg/dL), consistent with immune dysregulation associated with AT.
Regarding treatment, 68.4% of the patients received IVIG replacement therapy, 15.8% received antibiotic prophylaxis alone, and 15.8% received combined IVIG and antibiotic prophylaxis.
Malignancy developed in 3 patients (15.7%) during follow-up. Two patients were diagnosed with Hodgkin lymphoma at 11 and 13 years of age, presenting with cervical lymphadenopathy in the sixth and eighth years after the diagnosis of AT, respectively. The patient diagnosed at 11 years of age died during oncologic treatment at 12 years of age, whereas the patient diagnosed at 13 years of age remains under oncologic follow-up. Additionally, one patient developed hepatocellular carcinoma (HCC) in the eighth year of follow-up and was diagnosed at 13 years of age; this patient died 4 months after diagnosis despite treatment. The distribution of multisystem manifestations in patients with AT is shown in Table 3.
Distribution of Multisystem Manifestations in Patients with Ataxia-Telangiectasia

A total of 6 patients (31.5%) died during follow-up. Four deaths were due to infection-related respiratory failure at the ages of 7, 11, 16, and 18 years, while 2 deaths were attributable to malignancy. Overall survival was evaluated using Kaplan-Meier analysis (Fig. 1). The median overall survival was 108 months (SE 2.6 months; 95% CI, 103.0–113.0 months).
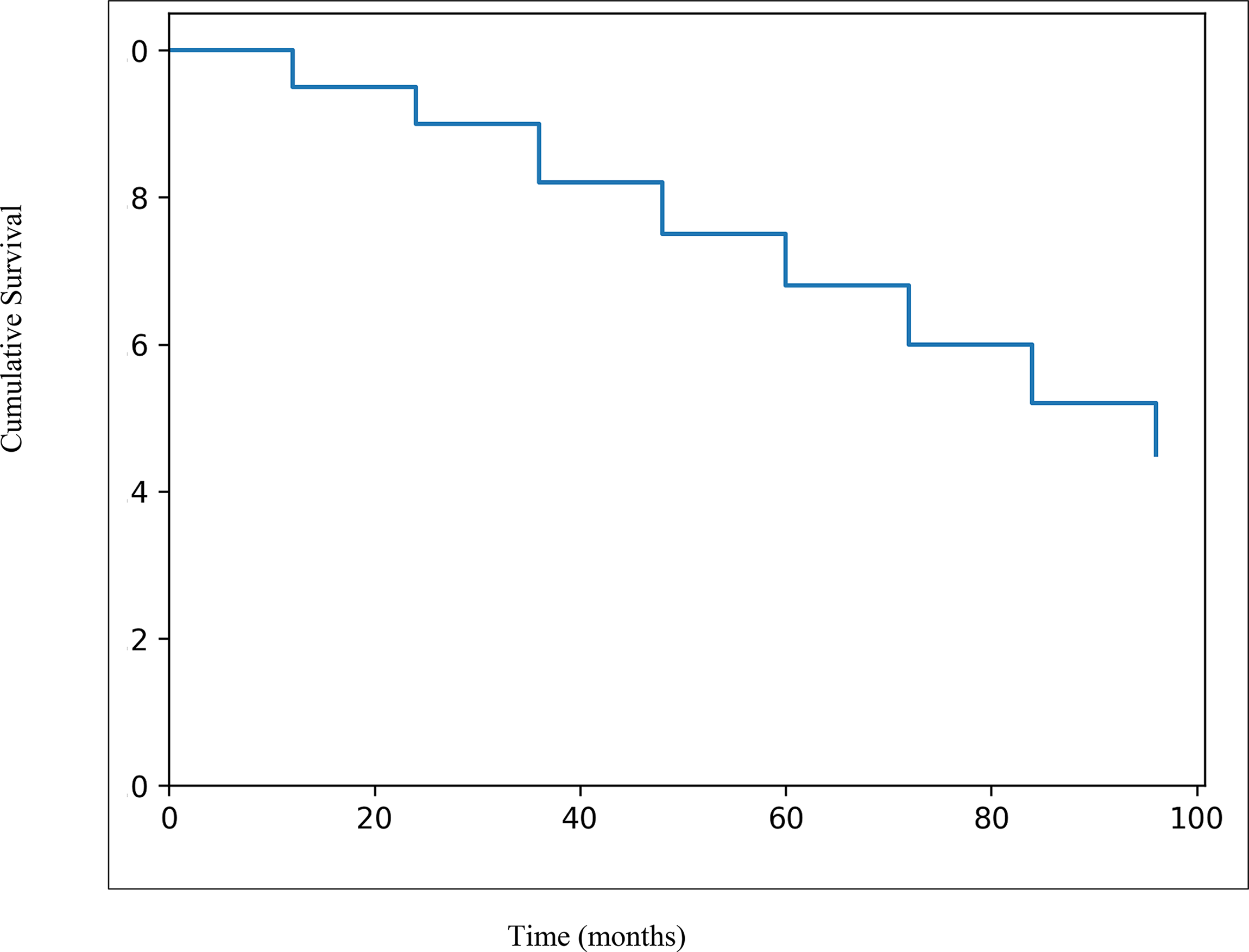
Kaplan–Meier overall survival curve of patients with ataxia-telangiectasia.
Discussion
This single-center cohort study evaluated the clinical characteristics, immunological profile, multisystem involvement, and outcomes of children with AT. The findings underscore the heterogeneous yet progressive nature of the disease, with neurological impairment representing the most consistent clinical feature.
Neurological ataxia was observed in all patients on physical examination. When presenting complaints were analyzed, gait disturbance recognized by family members was reported in 73.6% of patients. These findings are consistent with previous studies identifying early-onset cerebellar dysfunction as the hallmark of AT.4,5
Cutaneous manifestations in the present cohort included ocular telangiectasia, which was observed in all patients. Café-au-lait macules were detected in 31.5% of the patients. No other dermatologic findings were consistently observed in this study population.
Immunological involvement was reported in 56% of patients in a study of 91 pediatric cases published by Aktürk et al., IgA deficiency (41.8%) being the most frequently observed immunological defect. 19 In the study conducted by Çatal et al. in 25 children with AT, humoral immunodeficiency was present in all patients with immunological involvement. The most common deficiency was IgA (80%), followed by IgG (60%). Cell-mediated immunodeficiency was identified in 5 patients (20%). 20 In the present study, similar to the findings of Çatal et al., though at lower frequencies, humoral immunodeficiency was most commonly associated with IgA deficiency (57.9%), followed by IgG deficiency (15.8%). Among the 12 patients in whom IgG subclass levels were evaluated, IgG2 deficiency was detected in 75%.
Approximately 10% of patients with AT exhibit the Ataxia-telangiectasia with hyper-IgM syndrome (AT-HIGM) phenotype, characterized by reduced IgA and IgG levels with normal to elevated IgM levels.9,10 In the present cohort, 2 of 19 patients (10.5%) met the criteria for the AT-HIGM phenotype. IgM levels were above 2 SD in both patients. Genetic analysis revealed a homozygous ATM c.6047A > G (p.Asp2016Gly) mutation in one patient and a homozygous ATM c.4611 + 1 G > A mutation in the other. Although both variants have previously been reported in patients with AT, the homozygous c.6047A > G (p.Asp2016Gly) variant has not been associated with the hyper-IgM phenotype to date. Therefore, this study describes a novel clinical association between this ATM mutation and the AT-HIGM phenotype rather than a newly identified genetic variant.
Immune dysregulation phenotypes have been reported approximately 2-fold more frequently in patients with AT-HIGM compared with those with non-HIGM AT, with autoimmune hemolytic anemia being the most commonly described autoimmune manifestation. 10 In the present cohort, 1 of the 2 patients with AT-HIGM developed autoimmune hemolytic anemia accompanied by immune thrombocytopenia. No additional cases of autoimmune cytopenia were identified.
Consistent with previous reports, abnormalities in the cellular immune compartment were prominent in our cohort, particularly involving the CD3+, CD4+, CD8+, and CD19+ lymphocyte subsets. Similar findings have been widely reported in the literature, highlighting combined T- and B-cell involvement as a characteristic feature of AT.2,8 In a large multicenter study from Latin America, Pereira et al. demonstrated high frequencies of lymphopenia at the initial evaluation, with reductions observed in CD3+ T cells (76.5%), CD4+ T cells (79.5%), CD8+ T cells (55.0%), and CD19+ B cells (92.0%). 21 These findings underscore the early and profound impairment of adaptive immunity in affected patients.
Comparatively, Aktürk et al. reported lower rates of T-cell lymphopenia, with decreased CD3+, CD4+, and CD8+ lymphocyte counts observed in 26.4%, 35.2%, and 34.1% of patients, respectively, when evaluated using age-adjusted reference values. 19 The variability in reported frequencies across studies may reflect differences in patient age at diagnosis, disease severity, genetic heterogeneity, and timing of immunological assessment.
In this study, 63.1% of the 19 patients presented with frequent infections at diagnosis. Previous studies have reported high rates of recurrent sinopulmonary infections and chronic pulmonary complications, including bronchiectasis, in patients with AT.7,11,21 In our cohort, 5 patients (26.3%) developed bronchiectasis during follow-up.
Hepatic involvement, particularly hepatosteatosis, has been increasingly recognized as a metabolic complication in AT, although it has been reported in a limited number of studies. Previous cohorts have suggested that fatty liver disease may emerge during late childhood or adolescence, possibly related to chronic metabolic dysregulation and ATM-associated defects in insulin signaling pathways.12,13 In the present study, hepatosteatosis was identified in 26.3% of patients, with 1 patient diagnosed at presentation and 4 additional patients developing steatosis during follow-up. This finding supports growing evidence that hepatic steatosis represents a clinically relevant and potentially progressive component of AT.
Type 2 diabetes mellitus has been reported in 9.6% of patients in the Latin American AT cohort, with a median age at onset of 15.5 years. 21 In our cohort, 1 patient with hepatosteatosis subsequently developed type 2 diabetes mellitus, further supporting the concept that metabolic complications in AT may evolve over time and require long-term surveillance.
In a large cohort study involving 508 individuals with AT, primary malignancy was identified in 84 patients (16.5%), with hematological malignancies accounting for the majority of cases (74%), followed by solid tumors (26%). Among patients younger than 18 years, non-Hodgkin lymphoma was the most frequently observed malignancy (57%), whereas solid tumors predominated in individuals aged 18 years and older (55%). The cumulative incidence of cancer reached 29% by 35 years of age. 16 In our cohort, malignancy developed in 3 patients (15.7%), with a mean age at cancer diagnosis of approximately 10 years; 2 of these patients were diagnosed with Hodgkin lymphoma. The age at malignancy diagnosis in our cohort is consistent with previously reported pediatric AT cohorts, in which hematological malignancies typically occur during childhood.16,19 These findings further support the established elevated cancer risk associated with AT.
One notable finding in our cohort was the development of HCC in an 8-year-old patient, despite solid tumors being more commonly reported in adulthood among individuals with AT. Recently, Alesaeidi et al. reported HCC in a 14-year-old male with AT, further supporting the possibility of early-onset HCC in this population. 22 Together with this recent report, our case adds to the limited literature and underscores the importance of heightened hepatic surveillance in children with AT.
In the multicenter study by Pereira et al., prophylactic antibiotics were administered to 57.7% of patients, while 49.1% received immunoglobulin replacement therapy. 21 In contrast, a higher proportion of patients in our cohort received IVIG replacement therapy, whereas antibiotic prophylaxis was less frequently used. This difference may reflect center-specific treatment strategies, differences in infection burden, or earlier initiation of immunoglobulin replacement, potentially reducing the need for long-term antibiotic prophylaxis.
AT is a complex multisystem disorder associated with significant immunological heterogeneity and variable clinical outcomes. The findings of this study highlight the high frequency of humoral immune defects—particularly IgA deficiency—the occurrence of the AT-HIGM phenotype, and the substantial burden of infectious and pulmonary complications in affected children. These results underscore the importance of early immunological assessment, regular malignancy surveillance, and proactive management of sinopulmonary infections to improve long-term outcomes.
Authors’ Contributions
S.Ç.: Writing—original draft (lead); Formal analysis (lead); Writing—review and editing (equal). F.G.: Conceptualization (lead); Writing—original draft (equal); Formal analysis (lead); Writing—review and editing (equal). N.G.: Software (lead); Writing—review and editing (equal). S.Ö.B.: Methodology (lead); Writing—review and editing (equal). İ.A.H.: Conceptualization (supporting); Writing—original draft (supporting); Writing—review and editing (equal). İ.T.: Writing—review and editing (equal). Ö.A.: Writing—review and editing (equal). M.Ş.K.: Writing—review and editing (equal).
Footnotes
Financial Disclosure
The authors declared that this study has received no financial support.
Author Disclosure Statement
No conflict of interest was declared by the authors.
Funding Information
No funds, grants, or other support was received to conduct this research.