
Abstract
Background:
Diaphanous-related formin 1 (DIAPH1) is a cytoskeletal protein belonging to the formin family. Homozygous loss-of-function variants in DIAPH1 have been associated with seizures, cortical blindness, mitochondrial dysfunction, and combined immunodeficiency. Here, we report a rare case of homozygous DIAPH1 deficiency presenting with the classical clinical features of the disorder but without cortical blindness.
Case Presentation:
An 11-year-old male patient presented with a productive cough. His computed tomography scan revealed diffuse bronchiectasis and chronic parenchymal infiltrative lesions. Respiratory examination revealed bilateral crackles. Ophthalmologic examination and visual field testing were normal. Laboratory evaluation showed elevated serum IgG and IgE levels, whereas the IgA level was within the normal reference range and the IgM level was decreased for his age. Vaccine responses to hepatitis B and rubella were adequate; however, the isohemagglutinin titer against the blood group was insufficient. Lymphopenia was detected in the complete blood count. Although CD3+, CD4+, and CD8+ T cell, B cell, and natural killer cell counts were within normal ranges, class-switched memory B cells and recent thymic emigrants (CD4+CD45RA+CD31+) were significantly reduced. Next-generation sequencing-based targeted gene panel analysis, performed with a preliminary diagnosis of immunodeficiency, identified a homozygous nonsense variant, c.1051C > T (p.Arg351*), in exon 11 (11/15) of the DIAPH1 (NM_005219) gene. Epstein–Barr virus (EBV)-associated classical Hodgkin lymphoma subsequently developed in the patient, and chemotherapy was initiated.
Conclusions:
This is the first case of homozygous DIAPH1 deficiency presenting without cortical blindness. Homozygous DIAPH1 deficiency may present with a broad clinical spectrum and phenotypic variability, highlighting an increased susceptibility to EBV infection and virus-associated malignancies. Comprehensive immunological evaluation and close, long-term follow-up are essential for affected patients.
Case Presentation
An 11-year-old boy presented to the hospital for elective circumcision. During the preoperative evaluation, he was noted to have a productive cough. Physical examination revealed diffuse bilateral rales, and chest radiography demonstrated bilateral infiltrates. Consequently, he was admitted to the pediatric ward with a diagnosis of pneumonia. His medical history was notable for recurrent otitis media beginning at 3 months of age and hospitalizations at least twice annually for lower respiratory tract infections. Four years prior, he had been admitted to the pediatric intensive care unit with suspected encephalopathy characterized by blurred vision and convulsions. There was also a history of seizures during early childhood. Upon admission, the patient continued to exhibit a productive cough. Chest imaging revealed bronchiectatic changes, chronic parenchymal abnormalities, and an infiltrative lesion extending to the basal region of the right lung. On physical examination, his height and weight were below −3 standard deviation (SD), and his head circumference was below −6 SD. Dysmorphic facial features were noted. Diffuse verrucous papillomatous lesions were present on the arms, legs, and trunk, along with digital clubbing (Fig. 1). A Bacille Calmette–Guérin vaccination scar was observed. Bilateral diffuse rales were detected on auscultation. The patient exhibited age-inappropriate intellectual functioning; although speech was preserved, cognitive processing and comprehension were impaired. Neurological examination demonstrated preserved motor function, and gross motor development was appropriate for his age. Visual evoked potentials, visual acuity testing via a Snellen chart, and optic nerve examination performed by a pediatric ophthalmologist revealed no pathological findings.
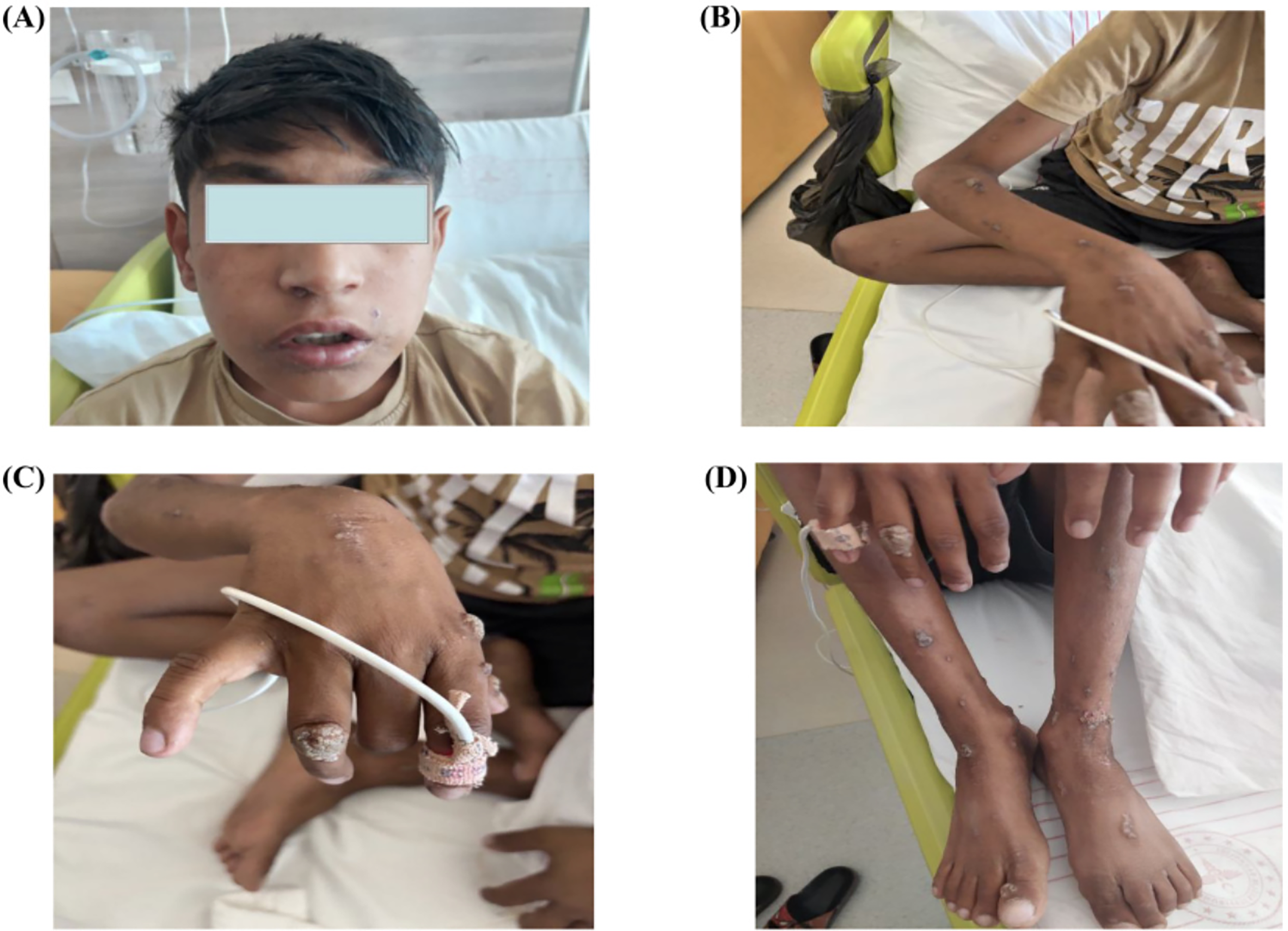
Dysmorphic facial appearance of the patient
The patient had three healthy siblings. His parents were first cousins, and there was no family history of a similar disease or primary immunodeficiency.
Laboratory findings are summarized in Table 1. At admission, the absolute lymphocyte count was 1,400 cells per microliter. Previous complete blood counts performed 3 weeks and 6 months prior showed lymphocyte counts of 870 and 1,280 cells per microliter, respectively. Serum IgG and IgE levels were mildly elevated, whereas the IgA level was within normal limits and the IgM level was slightly decreased for his age. Vaccine responses to hepatitis B and rubella were adequate. The patient’s blood group was B Rh-positive, and the anti-A isohemagglutinin titer was 1:4.
Immunophenotyping Results with Age-Matched Reference Ranges
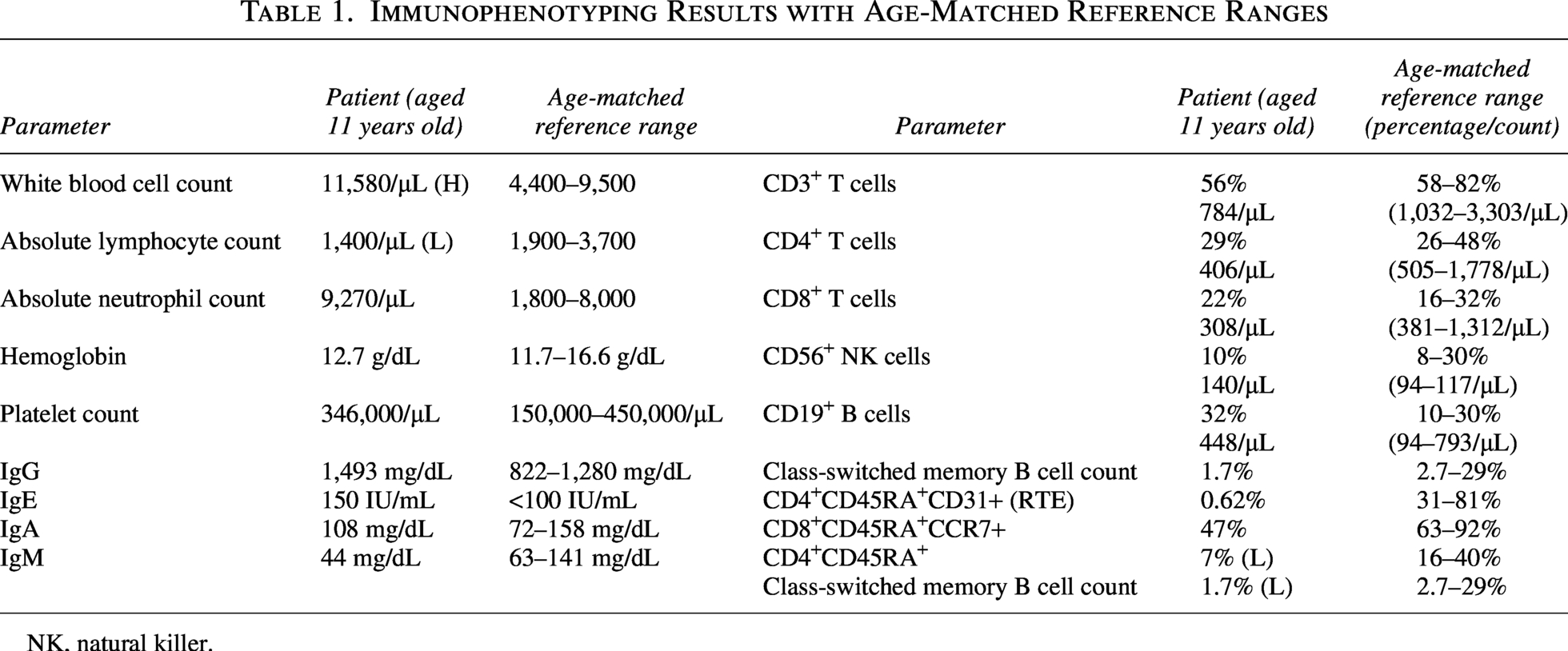
NK, natural killer.
Flow cytometric analysis demonstrated decreased CD3+, CD4+, and CD8+ T cell counts. Although B cell and natural killer (NK) cell counts were within normal ranges, class-switched memory B cells were markedly reduced. Recent thymic emigrants (CD4+CD45RA+CD31+) along with naive CD8+ and CD4+ T cells were significantly decreased.
Viral PCR testing was positive for Epstein–Barr virus (EBV), whereas PCR tests for cytomegalovirus and human immunodeficiency virus PCR tests were negative. Liver function tests revealed (AST: 38 IU/L, ALT: 88 IU/L, ALP: 799 IU/L, GGT: 44 IU/L). Renal function tests were normal.
Thoracic computed tomography (CT) revealed multiple areas of bronchiectasis (Fig. 2). Cranial magnetic resonance imaging (MRI) was unremarkable. However, cranial CT demonstrated an increased cerebrospinal fluid space surrounding both optic nerves, along with mild tortuosity. Abdominal ultrasonography was normal.
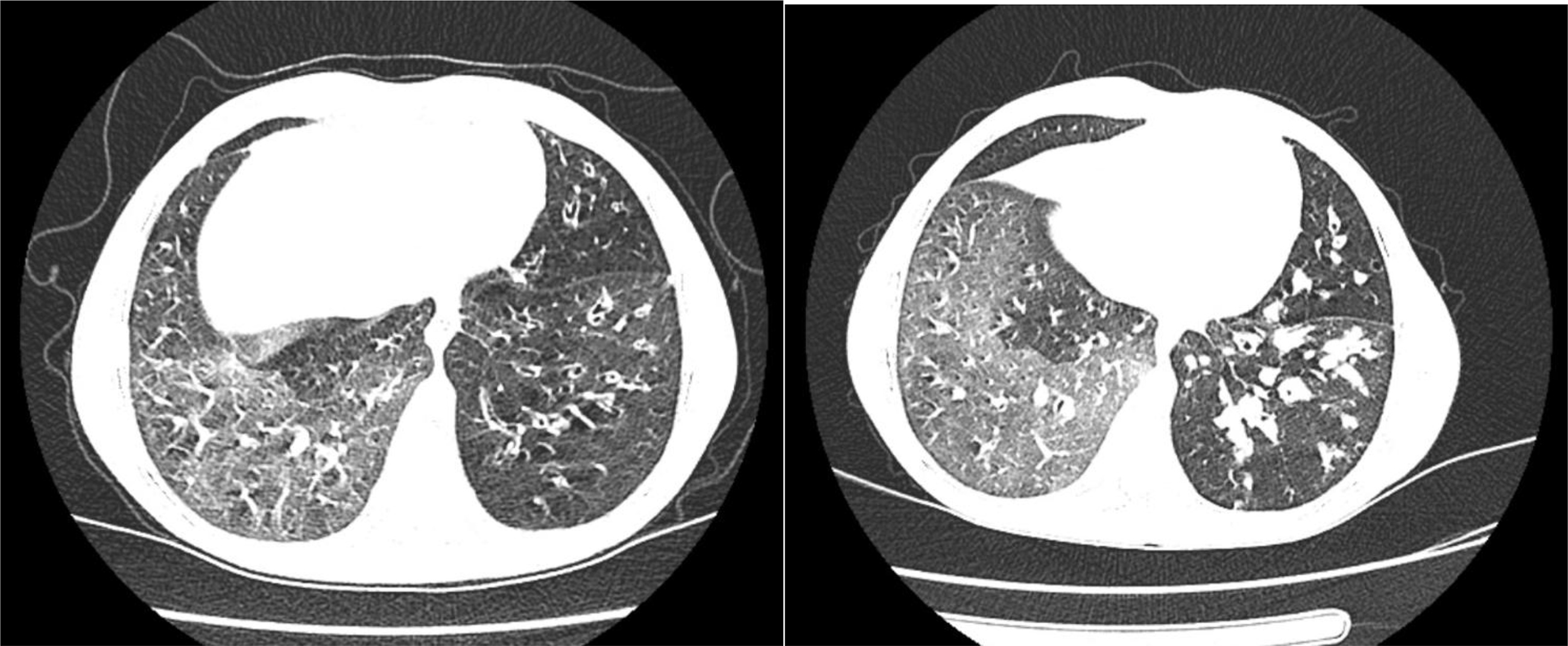
Chest computed tomography (lung window) demonstrating diffuse bilateral bronchiectatic changes.
Bronchoscopy revealed a normal subglottic region and trachea without structural abnormalities or foreign bodies; however, dense purulent secretions were diffusely present. Purified protein derivative skin testing showed an induration of 0 mm. No mycobacterial growth was detected in bronchoalveolar lavage cultures, and mycobacterial PCR testing was negative. Histopathological evaluation of the verrucous skin lesions was consistent with human papillomavirus infection.
As part of the diagnostic evaluation, an immunodeficiency expanded gene panel and Sangermsequencing were performed using molecular genetic methods. Genomic DNA was extracted from the patient’s peripheral blood sample following standard procedures. The sequencing library was prepared using the Clinical Exome Solution v3 capture kit (SOPHiA Genetics SA, Switzerland) and sequenced on the MiSeq platform (Illumina Inc., USA). The generated data were analyzed using current databases, including PubMed, OMIM, DGV, ClinVar, DECIPHER, and ClinGen; variant pathogenicity was assessed according to the American College of Medical Genetics and Genomics (ACMG) guidelines. 1 The identified variant was reported based on the NM_005219 transcript of the diaphanous-related formin 1 (DIAPH1) gene in the NCBI RefSeq database. For variants with the potential to explain the phenotype, segregation analysis was performed in the parents and siblings, and validation was carried out via Sanger sequencing using the Applied Biosystems 3500 Genetic Analyzer (Thermo Scientific, USA).
In next-generation sequencing-based targeted gene panel analysis performed with a preliminary diagnosis of immunodeficiency, a homozygous nonsense variant, c.1051C > T (p.Arg351*), was identified in exon 11 (11/15) of the DIAPH1 (NM_005219) gene. According to the ACMG guidelines, this variant was classified as pathogenic based on the evidence categories PVS1, PM2, PS4, and PP5. The variant had been previously reported as pathogenic in the ClinVar database (accession: VCV001990251.5). The presence of the homozygous variant in the patient was validated via Sanger sequencing. Segregation analysis performed in the healthy mother, father, and two siblings demonstrated that they carried the variant in the heterozygous state (Fig. 3).
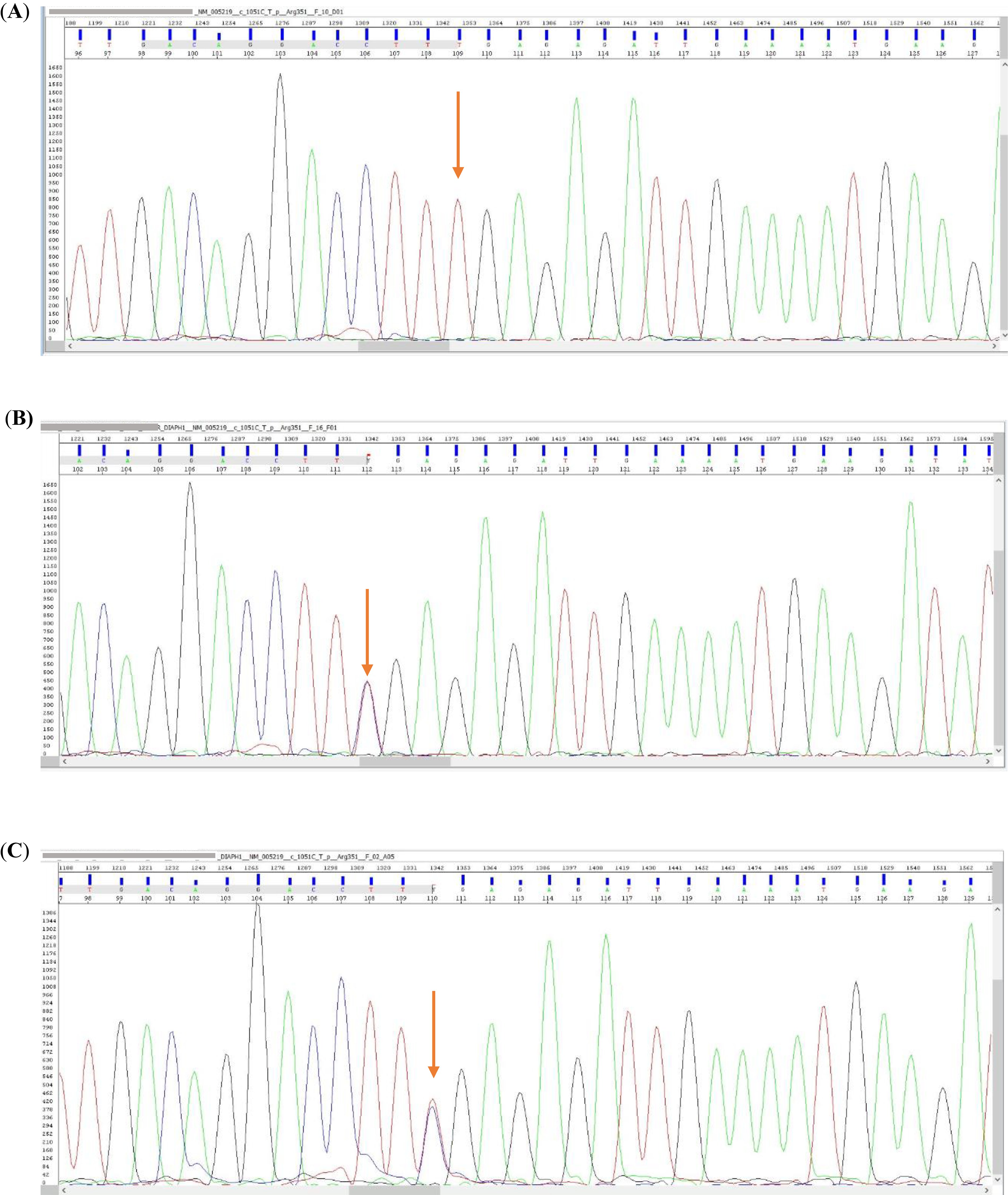
Segregation analysis of the proband
Following the diagnosis of homozygous DIAPH1 deficiency, monthly intravenous immunoglobulin replacement therapy and trimethoprim–sulfamethoxazole prophylaxis were initiated.
During follow-up, a 5 × 2 cm lymphadenopathy was detected in the right infra-auricular region. Ultrasonography revealed a heterogeneous lesion with cystic components. An incisional biopsy confirmed classical Hodgkin lymphoma, nodular sclerosis subtype. The patient subsequently received two cycles of the ABVD (adriamycin, bleomycin, vinblastine, and dacarbazine) regimen. Although EBV had initially been detected in the lymph node tissue, the patient’s EBV viral load became negative during the follow-up period.
He has a healthy brother who is a fully matched (10/10 human leukocyte antigen tissue type) donor without any genetic mutations. Consequently, preparations for bone marrow transplantation are currently underway. Informed consent was obtained from the patient’s parents for the publication of this case report and any accompanying images.
Discussion
Autosomal recessive DIAPH1 deficiency was first described in humans in 2015. 2 DIAPH1 is one of the 15 human formins and plays a central role in cytoskeletal remodeling by regulating actin filament polymerization and elongation.1–4 To date, all reported cases have involved homozygous mutations confirmed by genetic testing.5–7 Clinically, DIAPH1 deficiency was initially described within the spectrum of seizure, cortical blindness, and microcephaly syndrome (SCBMS); however, subsequent reports have expanded the phenotype to include combined immunodeficiency.3,7 Affected patients exhibit decreased T cell counts, profound depletion of naive CD4+ and CD8+ T cells, impaired regulatory T cell (Treg) differentiation, NK cell cytotoxic dysfunction, and defects in IL-2/STAT5 signaling. Consistent with these findings, our patient demonstrated reduced CD3+, CD4+, and CD8+ T cell counts, along with impaired Treg differentiation and susceptibility to EBV. Actinopathies (such as DOCK8 deficiency) are known to cause cutaneous viral susceptibility and cytoskeletal defects in epithelial immunity. Defective immune synapse formation is an observed phenotype of DIAPH1 deficiency; DIAPH1, a member of the formin family, is highly expressed in T cells and is required for TCR-mediated signal transduction. Although ZAP70 phosphorylation occurs normally following TCR stimulation, LAT phosphorylation depends on formin-mediated actin polymerization. Loss or inhibition of DIAPH1 function disrupts this process and impairs downstream LAT phosphorylation. Consequently, the immunological phenotype of DIAPH1 deficiency may partially or closely resemble that of LAT deficiency. 3 Moreover, DIAPH1 deficiency has been associated with impaired NK cell function and reduced cytotoxic activity. 3 This was demonstrated in a recent study examining T cells, NK cells, and helper ILCs in six unrelated patients with homozygous loss-of-function mutations in DIAPH1, as well as the consequences of shRNA-mediated silencing of DIAPH1 in Jurkat T cells. 3 Analysis of the TCR signaling pathway revealed impaired TCR signaling components, including ZAP70 and NF-kappa-B, after the knockdown of DIAPH1 in Jurkat cells upon stimulation with anti-CD3/CD28.
Beyond its immunological manifestations, DIAPH1 deficiency also affects neurological and mitochondrial functions. Given its role in cytoskeleton–mitochondrial tethering, affected individuals may develop mitochondrial dysfunction and respiratory chain complex IV deficiency, further compromising immune cell metabolism and function. In addition, DIAPH1 contributes to key processes in brain development, including neuronal migration and neurite formation.7,8 Notably, cranial MRI in our patient was unremarkable. Two patients without cortical blindness have been reported by Azizoglu et al.; these previously described cases carried the same mutation as our patient but presented with significant neurological involvement, including seizures, microcephaly, and developmental delay. One of those cases had concomitant autoimmune hemolytic anemia and subsequently died. 3
Management of DIAPH1 deficiency remains challenging; neurological manifestations can be refractory, and immunological complications are often severe. Patients appear particularly susceptible to uncontrolled EBV infection and EBV-associated lymphoproliferative disease.5,9 DIAPH1 is critical for neurodevelopment, immune regulation, and DNA repair. The underlying DNA repair defect may influence susceptibility to infections, lymphoma, or treatment-related toxicity. 10 In a recent article, two patients with DIAPH1 deficiency caused by different mutations passed away due to Hodgkin disease. This predisposition to developing lymphoid malignancies may increase not only because of reduced T cell function and impaired immunity against common B cell-tropic oncogenic viruses like EBV but also because the underlying DNA repair defect may serve as a contributory factor. 10 The development of EBV-associated classical Hodgkin lymphoma in our patient further supports this association. Biallelic mutations in CD27 or CD70 typically present with chronic EBV viremia, Hodgkin lymphoma, and/or hypogammaglobulinemia. Such patients also suffer from recurrent bacterial and viral infections, underlining the critical role of CD27–CD70 interactions in host defense beyond EBV immunity. 11 A recent study mentions that the development of Hodgkin lymphoma could also be caused by a defect in DIAPH1-dependent DNA double-strand break repair. 12
Current evidence supports redefining DIAPH1 deficiency as a disorder characterized by both SCBMS and primary immunodeficiency with immune dysregulation. The underlying pathogenesis is largely attributable to defects in cytoskeletal organization and mitochondrial function, leading to impaired immune synapse formation, defective TCR signaling, reduction of the naive T cell pool, and dysfunction of NK and innate lymphoid cells. These findings indicate that the loss of DIAPH1 has broad effects on both adaptive and innate immunity.6,7
In conclusion, our case expands the clinical spectrum of homozygous DIAPH1 deficiency by demonstrating that cortical blindness is not an obligatory feature. Our findings underscore the phenotypic variability of the disorder and highlight an increased susceptibility to EBV infection and virus-associated malignancies. Comprehensive immunological evaluation and close, long-term follow-up are essential for affected patients.
Ethical Statement
As this article is a case report, the patient and his parents gave permission.
Footnotes
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
No funding was received for this article.