
Abstract
During oncology drug development, it is important that ethnic differences are evaluated to determine the optimal dose and administration schedule in a new region based on the clinical data from other regions. The objective of this study was to explore the possibility of detecting ethnic differences in toxicity during early-phase clinical trials. Data were reviewed from phase I clinical trials for new drug applications conducted in Japan and Western countries. The maximum tolerated doses (MTDs), recommended phase II doses (RP2Ds), and approved doses in Japan were compared with those in Western countries. There were 4 of 28 drugs eligible for analysis that showed differences in MTDs or RP2Ds between Japanese and Western patients. Differences in MTDs or RP2Ds in 2 phase I trials were associated with ethnic differences in toxicity. It may be worthwhile to evaluate ethnic differences in toxicity during early-phase clinical trials for oncology drugs.
Introduction
Differences in the dosage and dose regimen of some drugs among regions have been pointed out, although they cannot definitely be attributed to ethnic differences. 1,2 Examination of ethnic differences is important while planning and conducting global clinical trials and determining whether clinical data from other countries or regions are applicable for clinical development in new countries or regions.
In the evaluation of ethnic differences during drug development, endogenous factors such as race, sex, and genetic polymorphisms and exogenous factors including socioeconomic factors and health care environments should be considered. 3 Examples of ethnic differences with known causes include those due to genetic polymorphisms in enzymes involved in drug metabolism and the ethnic differences in the distribution of these polymorphisms. In the development of S-1, differences in the distribution of the CYP2A6 polymorphism between Japanese and Western individuals caused different toxicity profiles, leading to differences in the maximum tolerated dose (MTD) and the recommended dose for subsequent clinical trials. 4 For irinotecan, variations in the distribution of the UGT1A1*6 and *28 polymorphisms by ethnicity resulted in different metabolism profiles, which resulted in different levels of toxicity. 5 An example of ethnic differences of unknown cause is the difference in the incidence of interstitial lung disease (ILD) with the use of gefitinib and bortezomib. The incidence of ILD is higher in Japanese patients than in Western counterparts. 6 –8 Ethnic differences in safety often pose a serious problem in the development of oncology drugs with narrow therapeutic windows.
If ethnic differences in the incidence of serious adverse events can be predicted early in drug development in a new region or country, it could be determined early on whether clinical data in other regions or countries can be used or whether additional data are necessary, and then clinical development would proceed more appropriately. For example, the development of erlotinib, which targets EGFR in the same manner as gefitinib, was based on information of an ethnic difference with a similar drug—that is, a higher incidence of ILD in Japanese patients with the use of gefitinib. Since this higher incidence was recognized in Japan, studies evaluating safety in Japanese persons were conducted during the development of erlotinib. 9,10 In addition, postmarketing data collection for erlotinib focused on the occurrence of ILD. 11 The clinical development of drugs in new countries or regions will proceed more appropriately if the extent of ethnic differences can be evaluated in an exploratory manner during phase I clinical trials that are first conducted in the residents of the new country or region, in addition to referring to the data on similar drugs.
In the present study, we examined the MTD in phase I clinical trials and the recommended phase II doses (RP2D) and approved doses of new oncology drugs to evaluate whether or not ethnic differences in toxicity can be detected in early-phase clinical trials in new countries or regions.
Methods
We reviewed the data from phase I clinical trials for new drug applications conducted in Japan and Western countries that had been reviewed by the Pharmaceutical and Medical Devices Agency (PMDA) and approved by the Japanese Ministry of Health, Labour, and Welfare between September 1999 and March 2011. Specifically, we examined the PMDA review reports—the documents submitted by the application sponsors, which have been publicly released on the websites of the PMDA 12 —and the published study reports to compare the MTD (or the maximum administered dose, if MTD was not reached) and the RP2D for the Japanese population and that in the US and Europe. The definitions of the terms in this study were as follows: MTD was the lowest dose level at which more than 33% of patients experience dose-limiting toxicity (DLT). RP2D was one dose level below the MTD.
To evaluate ethnic differences between Japanese and Western populations, we compared the approved doses of all drugs according to the prescribe information shown on the website of the regulatory agencies in each region, 13 –15 and we retrospectively analyzed the safety profile and frequency of adverse events of all drugs based on the published study reports when differences in MTD or RP2D were identified.
To assess the adequacy of phase I clinical trial design for detecting any differences in toxicity, we compared the dose escalation methods and reasons for stopping dose escalation in the Japanese trials with those conducted in the US and Europe.
No statistical comparisons were made because of the retrospective nature of this analysis.
Results
Between 1999 and 2011, a total of 97 oncology drugs were approved in Japan. Among them, 39 drugs with novel active ingredients were approved. The following drugs were excluded from this study: 4 drugs that had not been approved in the US and Europe (miriplatin, tamibarotene, talaporfin, amrubicin); 3 hormonal drugs (letrozole, exemestane, anastrozole); 2 drugs for which phase I clinical trials were not conducted in Japan (thalidomide, nelarabine); 1 drug for which dose escalation studies were not conducted in the US and Europe (azacitidine); and 1 drug used with different supportive therapies between Japan and the US and Europe (pemetrexed). Thus, 28 drugs were examined in this study.
Drugs With Differences in MTD, RP2D, and Approved Doses Between Japanese and Western Populations
Differences in MTD or RP2D between Japanese and Western populations were observed for 4 of 28 drugs: temsirolimus (with differences only in MTD) and capecitabine, fludarabine, and topotecan (with differences in both MTD and RP2D). Among the drugs with differences in MTD or RP2D, fludarabine and topotecan had different approved dosages and dose regimens. These differences and details of DLT are shown in Table 1. For the drugs without differences in MTD or RP2D, there was also no differences in the approved dosage and dose regimen.
MTD, RP2D, approved dose, and DLT of drugs with different toxicity profiles between Japanese and Western populations found in phase I trials.
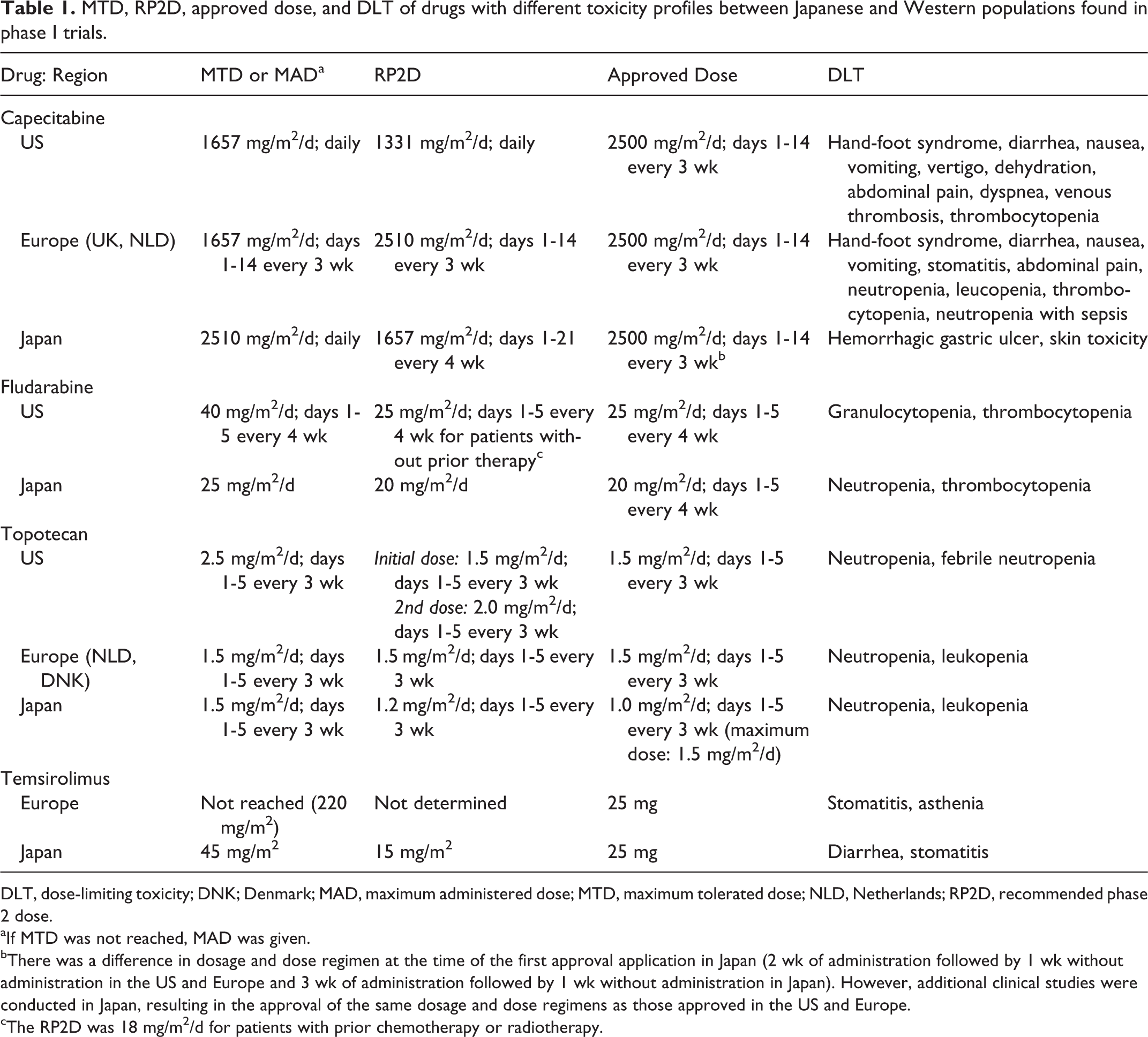
DLT, dose-limiting toxicity; DNK; Denmark; MAD, maximum administered dose; MTD, maximum tolerated dose; NLD, Netherlands; RP2D, recommended phase 2 dose.
aIf MTD was not reached, MAD was given.
bThere was a difference in dosage and dose regimen at the time of the first approval application in Japan (2 wk of administration followed by 1 wk without administration in the US and Europe and 3 wk of administration followed by 1 wk without administration in Japan). However, additional clinical studies were conducted in Japan, resulting in the approval of the same dosage and dose regimens as those approved in the US and Europe.
cThe RP2D was 18 mg/m2/d for patients with prior chemotherapy or radiotherapy.
Safety Profiles of the Drugs With Differences in MTD, RP2D, and Approved Doses
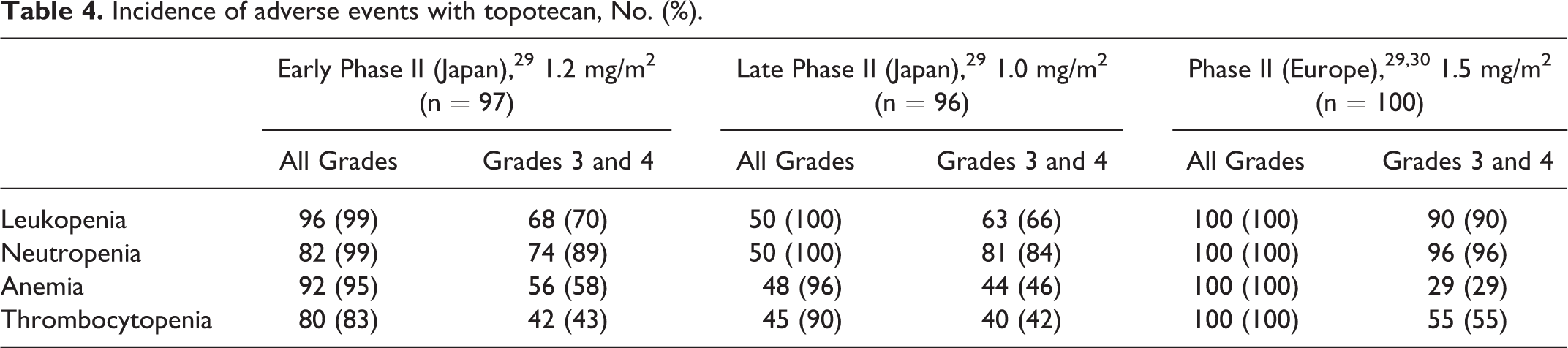
The incidence of adverse events with capecitabine—including pigmentation, diarrhea, increased aspartate aminotransferase level, and elevated bilirubin level—was different between Japanese and non-Japanese patients (Table 2). For temsirolimus, a higher incidence of stomatitis and ILD was observed in Japanese persons than in non-Japanese persons (Table 3). The safety profile of topotecan and fludarabine could not be compared owing to the lack of studies conducted using the same dose regimens in Japan as in the US or Europe. However, the occurrence rate of hematologic toxicity with topotecan in Japanese patients is the same as in European patients despite using their different doses, suggesting that there are differences in the occurrence rate of hematologic toxicity between Japanese and European patients (Table 4). Besides, a higher incidence of hematologic toxicity was observed with fludarabine at lower doses in Japanese people than in US people. The incidence of neutropenia was 69% in Japanese people and 18% in US people (Table 5).
Incidence of treatment-related adverse events with capecitabine (2500 mg/m2 for 14 d, every 3 wk), No. (%).
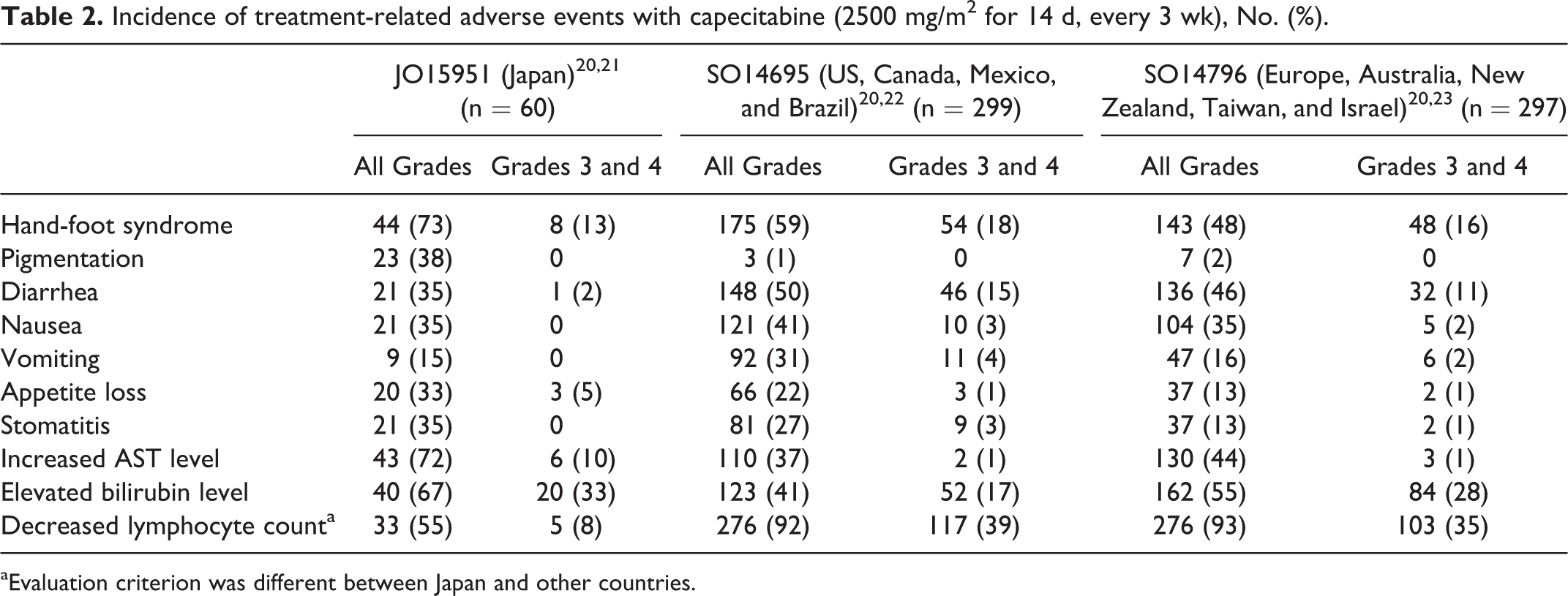
aEvaluation criterion was different between Japan and other countries.
Incidence of adverse events with temsirolimus (25 mg), No. (%).
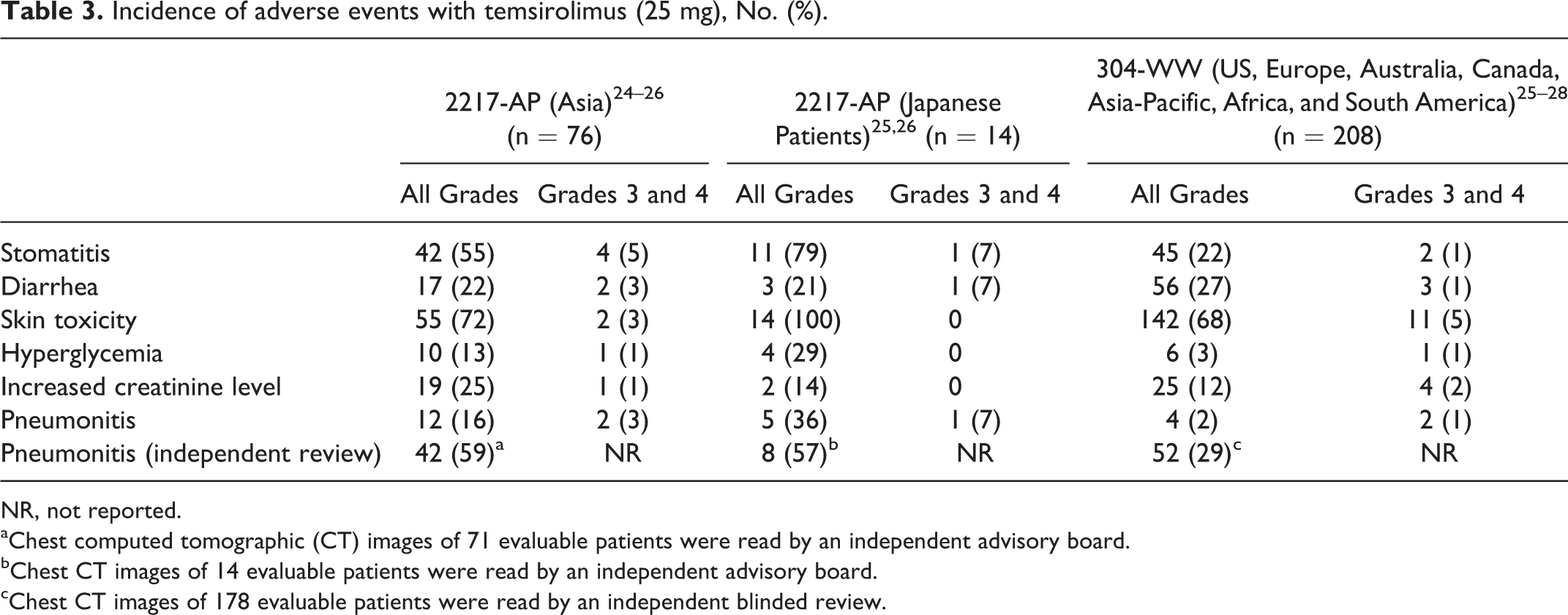
NR, not reported.
aChest computed tomographic (CT) images of 71 evaluable patients were read by an independent advisory board.
bChest CT images of 14 evaluable patients were read by an independent advisory board.
cChest CT images of 178 evaluable patients were read by an independent blinded review.
Incidence of adverse events with topotecan, No. (%).
Incidence of adverse events with fludarabine, No. (%).
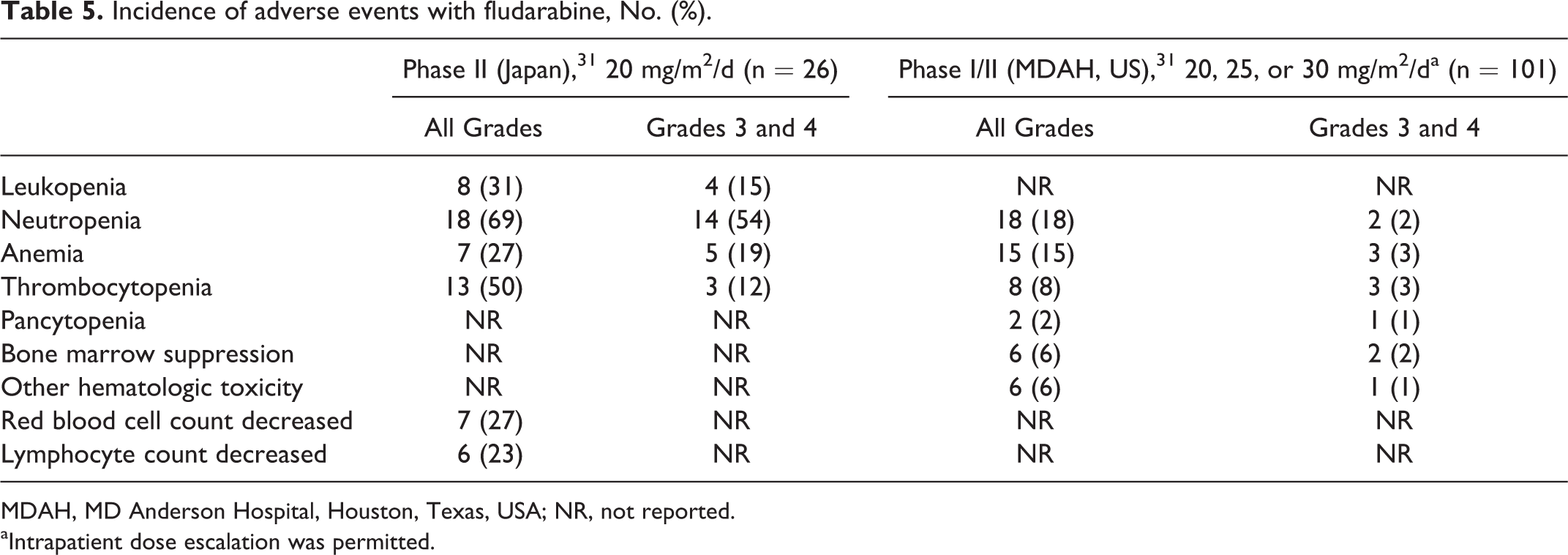
MDAH, MD Anderson Hospital, Houston, Texas, USA; NR, not reported.
aIntrapatient dose escalation was permitted.
Dose Escalation Methods and Reasons for Stopping Dose Escalation
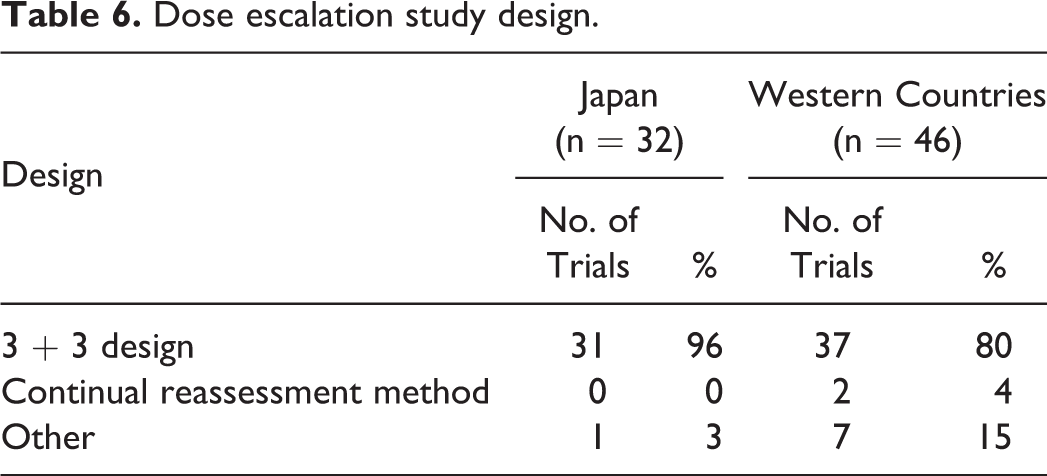
According to the PMDA review reports, for the 28 drugs examined, 78 dose escalation studies were conducted, which consisted of 32 studies in Japanese patients and 46 in European and American patients.
The dose was increased in a 3 + 3 design in 31 of 32 studies in Japanese patients and another design in the remaining study. In the 46 studies in Western persons, the dose was increased in a 3 + 3 design in 37 studies, with a continual reassessment method in 2 studies and other designs in 7 studies (Table 6).
Dose escalation study design.
In the 32 studies with Japanese participants, the reason for discontinuation of dose escalation was toxicity in 8 studies, confirmation of the tolerability of the overseas recommended dose in 20 studies, and other in 4 studies. In the 46 studies with Western participants, the reason was toxicity in 24 studies, consideration of pharmacokinetics in 3 studies, achievement of the dose expected to block the target in 3 studies, and other in 16 studies (Table 7).
Reason for stopping clinical trials.

aAlmost all studies had the objective of evaluating tolerability of the dosage approved for Western populations.
Discussion
In the present study, 2 cytotoxic drugs—fludarabine and topotecan—showed hematologic toxicity in phase I trials. This eventually led to different doses of these drugs being approved in Japan and in the US and Europe. We cannot confirm ethnic differences from the results of the phase I trials, as the number of patients was limited. However, this finding suggests a hypothesis that the differences in MTD or RP2D in early clinical trials may be associated with ethnic differences in toxicity. Therefore, when we found the differences in MTD or RP2D, we might need to collect additional data, including pharmacokinetics, genetic polymorphism, and other ethnic factors.
It is unclear why the approved doses of both fludarabine and topotecan were different in 2 regions. These drugs had a DLT, which was hematologic toxicity. However, the other drugs with hematologic toxicity as the DLT did not have different approved doses.
In the pharmacokinetic study of fludarabine, the area under the curve of plasma 2F-ara-A, which is the active metabolite of fludarabine phosphate, was similar between the Japanese and American patients. 16 Although the distribution of the common variant alleles of CYP genes is known to vary among different ethnic populations, 17 in an in vitro study, 3H-2F-ara-A was not metabolized by CYP3A4 and CYP1A2.
Topotecan is a topoisomerase I inhibitor, which is a water-soluble derivative of camptothecin. The pharmacokinetic parameters with topotecan—Cmax, area under the curve, and T1/2 levels in the plasma—were not different between Japanese and Western patients. Human liver microsomal metabolism of topotecan and its metabolite was not affected by CYP1A2, CYP2A6, CYP2C8/9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP4A. 17
Yet, capecitabine and temsirolimus showed no differences in approved dosages and dose regimens, although both MTD and RP2D were different between Japan and the US and Europe. For temsirolimus, although gastrointestinal toxicity such as diarrhea and stomatitis was caused by a considerable disparity in MTD between Japan and Europe, the safety profile of this drug in later clinical trials showed a difference in the incidence of not only stomatitis but also ILD.
The reason for such discrepancies in ethnic differences between earlier and later clinical trials is unknown. Note that observed ethnic differences in early clinical trials can be attributed to patient-level differences because these studies were conducted with the limited number of patients. Examination of study designs in the present study showed that 20 of 32 phase I clinical trials in new regions (62.5%) did not employ a dose escalation design to determine a region-specific MTD but rather attempted to confirm tolerability of the doses recommended in the regions where the drugs were previously developed. Only 8 studies (25%) specifically evaluated the development of toxicity to determine MTD. Therefore, we speculate that even if tolerability in new regions is similar to that in the previously approved regions, dosages and dose regimens in new regions would not be sufficiently evaluated.
In the present study, as far as we can determine, the approved doses were the same for drugs without any differences in MTD or RP2D between Japanese and Western participants in early clinical trials. However, some drugs without any differences in MTD or RP2D demonstrated different toxicity profiles in Japanese participants. For example, there was a higher incidence of ILD with gefitinib and bortezomib in Japanese participants. 6 –8
Two theories have been put forth on why there was a failure to detect ethnic differences in the toxicity profile of gefitinib and bortezomib in early clinical trials and a discrepancy between the toxicity observed in the early versus later clinical trials for temsirolimus and capecitabine. First, less frequent adverse events cannot be detected in clinical studies with a small number of participants. In the present study, the toxicity of the 2 drugs (fludarabine and topotecan) shown to be different among different populations was hematologic toxicity, a relatively frequent adverse event. Conversely, early clinical trials with the small number of participants have only a limited capacity to detect ethnic differences in adverse events with relatively low incidences—for example, ILD. Depending on the properties of the specific drug and those in the same class, it may be more helpful to search for evidence of ethnic differences in later clinical trials. Second, dose escalation design was not strictly followed in the phase I trials when a drug is being studied for a new region. For bortezomib, dose escalation was discontinued because tolerability of the overseas recommended dose was confirmed and a sufficient determination of MTD was not performed.
A potentially more significant problem is the possibility that uncommon but severe adverse events do not surface during the clinical development stage. In Japan, immediately after the launch of gefitinib, ILD associated with the drug’s use caused multiple cases of death. Its prescribing information was ultimately revised to raise awareness of the risk of ILD. 18,19 It should be recognized that information collected by early and late clinical trials is not sufficient. We consider it meaningful to collect the data from multinational trials, including early clinical trials, and continue the examination for ethnic differences in a larger number of patients, including postmarketing surveys.
Two limitations of the present study should be considered. One is that it examined only drugs that were eventually approved. Drugs whose development was discontinued, potentially due to ethnic differences detected during clinical development, were not examined. The other is that we could not find the information on the difference of sampling interval for laboratory variables and the criteria in each trial to report the laboratory-related adverse events between Japan and Western countries. As for the adverse event reporting, the slightly abnormal laboratory values tend to be strictly reported as adverse events in Japan, while they did not tend to in Western countries. Although these tendencies could not cause the ethnic differences in severe hematologic toxicity, these points should be noted in the interpretation of the results.
The present study found that phase I clinical trials detected ethnic differences in the toxicity profile of 2 of 28 drugs examined, suggesting that it is important to collect additional data in later clinical trials when MTD or RP2D in a new region is different from that in previously approved regions.
Footnotes
This study was presented as part of the European Multidisciplinary Cancer Congress, September 23-27, 2011, Stockholm, Sweden. The views expressed are the result of independent work and do not represent the views of the Pharmaceuticals and Medical Devices Agency of Japan.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research [C-24500345 to S.M.] from the Ministry of Health, Labour and Welfare of Japan.