
Abstract
Clinical trials are designed to evaluate the efficacy, safety, or other characteristics associated with medical products. Trials are usually complex and require a large group of professionals to follow a clinical trial protocol, standard operating procedures, and study-specific manuals, guidelines, and plans. Clinical trial protocols prospectively describe the background and rationale for conducting the trial, the objectives of the trial, the trial design, the equipment to be used, the procedures to be performed, and the statistical methods on how the trial data are to be analyzed. Deviations from the protocol can result in harm to subjects, biased or inaccurate results, and possible rejection of all or part of the trial data by the sponsor or regulatory authorities. Despite preventive efforts, protocol deviations are likely to occur in most trials. This position paper proposes a common definition of protocol deviations and recommends best practices for their detection, classification, and management as part of their life cycle, with a goal of reducing their impact on subject safety and data integrity. The information contained herein is drawn globally from industry experts within the DIA Good Clinical Practice and Quality Assurance community, an industry-wide survey, and presentations with discussions at various industry meetings.
Keywords
Introduction
For decades, the most frequent Food and Drug Administration (FDA) clinical trial enforcement actions (including 483 citations and warning letters) have been due to failures to follow the investigational plans. 1 In particular, these included departures from the clinical trial protocol or standard operating procedures (SOPs) that have had or may have had an adverse effect on the rights, safety, or well-being of subjects or a negative impact on the integrity of data collected to draw conclusions related to the objectives of the study.
The following is an excerpt from an FDA warning letter issued to an investigational site: 2. You failed to conduct the studies or ensure they were conducted according to the investigational plan, and failed to protect the rights, safety, and welfare of subjects under the investigator’s care [21 CFR 312.60].… a. As discussed in Item 2.i. below, Subject [redacted] had a Week 36 ECG conducted on July 16, 2009, that resulted in a QTcF value of 526 ms, according to the ECG machine readout. You failed to follow the protocol because you did not admit the subject to a telemetry unit or an emergency unit with continuous ECG recording capabilities until normalization of the QTcF interval.… Subject [redacted] had screening labs performed on September 22, 2008, that reported an AST of 63 U/L and an ALT of 87 U/L. These values are greater than 1.5 times the upper normal limit values of AST … and therefore render the subject ineligible to enroll in the clinical investigation.… You failed to follow the protocol by enrolling this subject in the clinical investigation. Furthermore, you failed to protect the rights, safety, and welfare of this subject and placed this subject at risk by maintaining this subject in the clinical investigation for the entire 52-week investigative period.
2
Avoidance of such deviations is ideal, but when they occur, the clinical trial team needs procedures and tools to track and effectively correct the situation and prevent it from happening again. There is currently no clear guidance from regulators or consistency in terminology across pharmaceutical companies to detect, assess, classify, or report protocol deviations. Therefore, in an effort to address definitions, classification, management, and reporting of protocol deviations, the DIA Good Clinical Practice and Quality Assurance community collaborated with worldwide industry experts to develop this position paper. In addition to input from DIA members, a survey was administered to industry professionals through various organizations, and feedback was obtained at several industry meetings. Based on feedback received from reviewers and at industry meetings, key issues were further discussed and the paper updated. Results from the survey are presented in Appendix 1.
The goal of this project was to address the following objectives: Determine a common definition for protocol deviations Document the typical life cycle from prevention through archival of protocol deviations Establish best practices for classifying and managing protocol deviations
Background
Protocol deviations can occur at clinical trial sites, at the sponsor (including contract research organizations [CROs] with delegated sponsor responsibilities), or at other vendors (including drug suppliers and laboratories). The scope of this paper is protocol deviations that originate at clinical trial investigational sites and at the sponsor, since the protocol primarily describes procedures to be followed at the sites and oversight of the trial conduct is the primary responsibility of the sponsor. Even though this position paper does not explicitly address deviations by other vendors involved in a trial, adherence to study-specific procedures by vendors is critical, and the process to detect, track, and manage deviations from planned procedures should be part of every company’s corrective action preventive action (CAPA) program. A thorough knowledge and understanding of the requirements of the protocol by all those involved, including trial subjects, are necessary to minimize the occurrence and impact of protocol deviations.
The US Health and Human Services Secretary’s Advisory Committee on Human Research Protections (SACHRP) held a meeting on February 28 and 29, 2012, the minutes of which made the observation that “a problematic area in human subject protection is the wide divergence among institutions, sponsors, investigators and IRBs [institutional review boards] regarding the definition of and the procedures for reviewing protocol deviations.” 4
Scientifically sound and well-designed clinical trial protocols with input from all stakeholders are essential to address the objectives of the trial. Careful planning and conduct with adequate oversight ensure subject safety and data accuracy. The level of detail in the protocol should be specific enough to allow consistency across the trial but also provide flexibility when needed. Timing of trial-specific assessments and selection of eligibility criteria are a delicate balance of too little detail versus too much. Too general eligibility criteria or too wide visit windows may result in high variability in the data, which can in turn affect the results and their interpretation. This is particularly relevant when trial results are negative, since the outcome of the trial may not be due to the lack of drug effect but rather due to a poor study design resulting in too much variability to detect a treatment effect within the study sample size. Too specific detail (ie, very strict precise requirements regarding who needs to do what and when) can result in the trial not reflecting “real world” clinical practice, as well as causing too many protocol deviations. Providing appropriate windows around dates or times of assessments allows for variability while still providing scientifically valid data within bounds. Additionally, if there are sponsor or vendor preferences in regard to methods, these should be expressly stated in the protocol, study manuals, guidelines, and plans, rather than implied or assumed.
Definitions
Deviations from protocol procedures happen in virtually all studies. These deviations are sometimes called “protocol deviations,” “protocol violations,” “protocol variances,” and “noncompliances.” The FDA Center for Device and Radiologic Health addresses protocol deviations in 21 CFR 812.150 (a) (4): Deviations from the investigational plan. An investigator shall notify the sponsor and the reviewing IRB (see §56.108(a) (3) and (4)) of any deviation from the investigational plan to protect the life or physical wellbeing of a subject in an emergency.… Except in such an emergency, prior approval by the sponsor is required for changes in or deviations from a plan, and if these changes or deviations may affect the scientific soundness of the plan or the rights, safety, or welfare of human subjects, FDA and IRB [approval] in accordance with §812.35(a) also is required.
5
A protocol deviation/violation is generally an unplanned excursion from the protocol that is not implemented or intended as a systematic change. A protocol deviation could be a limited prospective exception to the protocol (eg agreement between sponsor and investigator to enroll a single subject who does not meet all inclusion/exclusion criteria). Like protocol amendments, deviations initiated by the clinical investigator must be reviewed and approved by the IRB and the sponsor prior to implementation, unless the change is necessary to eliminate apparent immediate hazards to the human subjects (21 CFR 312.66), or to protect the life or physical well-being of the subject (21 CFR 812.35(a)(2)), and generally communicated to FDA. “Protocol deviation” is also used to refer to any other, unplanned, instance(s) of protocol noncompliance. For example, situations in which the investigator failed to perform tests or examinations as required by the protocol or failures on the part of study subjects to complete scheduled visits as required by the protocol, would be considered protocol deviations. Determine whether changes to the protocol were: Documented by an amendment, dated, and maintained with the protocol; Reported to the sponsor (when initiated by the clinical investigator); and Approved by the IRB and FDA (if applicable) before implementation (except when necessary to eliminate apparent immediate hazard(s) to human subjects).
6
III. B. 3. Protocol Violations that Present Unreasonable Risks There are occasions when a failure to comply with the protocol may be considered a failure to protect the rights, safety, and welfare of subjects because the non-compliance exposes subjects to unreasonable risks. For example, failure to adhere to inclusion/exclusion criteria that are specifically intended to exclude subjects for whom the study drug or device poses unreasonable risks (eg, enrolling a subject with decreased renal function in a trial in which decreased function is exclusionary because the drug may be nephrotoxic) may be considered failure to protect the rights, safety, and welfare of the enrolled subject. Similarly, failure to perform safety assessments intended to detect drug toxicity within protocol-specified time frames (eg, CBC for an oncology therapy that causes neutropenia) may be considered failure to protect the rights, safety, and welfare of the enrolled subject. Investigators should seek to minimize such risks by adhering closely to the study protocol.
7
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) E3 “Guideline for Industry: Structure and Content of Clinical Study Reports, Questions & Answers” defines protocol deviations as follows: A protocol deviation is any change, divergence, or departure from the study design or procedures defined in the protocol.
Important protocol deviations are a subset of protocol deviations that may significantly impact the completeness, accuracy, and/or reliability of the study data or that may significantly affect a subject’s rights, safety, or well-being. For example, important protocol deviations may include enrolling subjects in violation of key eligibility criteria designed to ensure a specific subject population or failing to collect data necessary to interpret primary endpoints, as this may compromise the scientific value of the trial.
Protocol violation and important protocol deviation are sometimes used interchangeably to refer to a significant departure from protocol requirements. The word “violation” may also have other meanings in a regulatory context. However, in Annex IVa, Subject Disposition of the ICH E3 Guideline, the term protocol violation was intended to mean only a change, divergence, or departure from the study requirements, whether by the subject or investigator, that resulted in a subject’s withdrawal from study participation. (Whether such subjects should be included in the study analysis is a separate question.) To avoid confusion over terminology, sponsors are encouraged to replace the phrase “protocol violation” in Annex IVa with “protocol deviation” as shown in the example flowchart below. Sponsors may also choose to use another descriptor, provided that that the information presented is generally consistent with the definition of protocol violation provided above. … The definition of important protocol deviations for a particular trial is determined in part by study design, the critical procedures, study data, subject protections described in the protocol, and the planned analyses of study data. In keeping with the flexibility of the Guideline, sponsors may amend or add to the examples of important deviations provided in E3 in consideration of a trial’s requirements. Substantial additions or changes should be clearly described for the reviewer.
9
The requirements and procedures described in the study protocol and protocol amendments (clinical investigational plan). Even if not explicitly mentioned in the protocol, these include adherence to: Good Clinical Practices (GCP) or other applicable Good Practices (GxPs) required for the conduct of the trial, and Legal and regulatory requirements applicable to the conduct of the trial.
We further propose that, as specified in ICH E3, 10 important protocol deviations be a subset of protocol deviations that may significantly affect the completeness, accuracy, or reliability of the study data or that may significantly affect a subject’s rights, safety, or well-being.
In recent years, there has been a trend to leave out details from the protocol and put them in study-specific plans. In general, deviations from these plans or SOPs would not be considered protocol deviations. However, any deviations from study plans or SOPs that may have a significant impact on the safety of study subjects or integrity of study data should be considered important protocol deviations.
The current lack of standards with use of the term “protocol violations” causes a lot of confusion. Different companies have created their own definitions with no consensus. Furthermore, per the ICH E3 Q&A guidance, the recommendation is to consider “protocol violation” equivalent to “important protocol deviations” when summarizing data in the clinical study report (CSR). Our recommendation is that, to avoid continued confusion, the term “protocol violations” no longer be used.
The Life Cycle and Management of Protocol Deviations
Anyone who has been involved with the conduct of clinical trials knows that despite everyone’s best intentions, protocol deviations do occur. We recommend that a trial-specific protocol deviations handling plan (PDHP) be developed and finalized prior to the first patient’s first visit in the trial. This plan should describe the approach for preventing, detecting, tracking, classifying, and following up on protocol deviations. This plan should be periodically reviewed and updated as new information becomes available. If a quality risk management plan is developed, the elements of the PDHP can be included in the plan. All key personnel involved in the trial—at the sponsor, CRO, and the site level—should be trained on these plans with regular oversight to ensure adherence. The training could be done at the investigator meeting or at the site initiation monitoring visit. The plan should also include measures to ensure consistency across systems that may record potential protocol deviations (such as monitoring reports, clinical data management/electronic data capture [CDM/EDC] systems, and clinical trial management systems [CTMSs]). The processes described in the plan should indicate responsibilities of individuals involved in the clinical trial who may detect a deviation—for example, study coordinators, site monitors, data reviewers, biostatisticians, medical monitors, and other personnel who review subjects’ data or analyses during the trial. Ongoing timely and periodic (eg, weekly or monthly) review of all protocol deviations in a study permit immediate actions and minimize the potential negative impact on subject safety or data integrity. As a result of such periodic reviews, the study team is aware of the overall number of deviations and any trends that may require an amendment to the protocol or retraining. In addition, it helps identify sites that have more deviations than others. The outcome and conclusions from these review meetings should be documented in writing.
The following stages constitute the life cycle of a protocol deviation: Preventive measures and a CAPA program Detection and timely reporting Tracking Classification Reporting in the CSR Archival
Stages (a) through (d) above are not necessarily discrete sequential steps but may happen iteratively or in parallel. They are listed separately for ease of describing them in detail. The following sections describe our recommendations for a standardized approach covering the life cycle of protocol deviations to be used by sponsors and CROs.
Preventive Measures and a CAPA Program
Protocol Complexity and Objectives
The key preventive measure to minimize protocol deviations is to develop a protocol that has been carefully reviewed by those involved in the operational aspects of conducting a trial. Protocols are often written by a few key authors who understand the therapeutic area, indication, and investigational product being tested but may not be familiar with the operational aspects of conducting the study at the site. Protocols that have input from site personnel who treat and evaluate patients regularly can provide guidance on how to minimize protocol deviations.
Clinical trial protocols continue to increase in complexity with multiple objectives and assessments included that add marginal benefit. Careful consideration should be given to the length of each assessment and its impact on trial subjects and site personnel. Too many complex assessments can tire subjects and site personnel, resulting in missed or inaccurate evaluations as well as subjects discontinuing from the study. There are ongoing efforts to standardize clinical trial protocols, and we suggest that readers refer to the “Standard Protocol Items: Recommendations for Interventional Trials,” or SPIRIT, initiative, 11 as well as the work underway by the Clinical Data Interchange Standards Consortium’s Protocol Representation Group. 12
Other methods to prevent deviations may include quality-by-design approaches, including describing select details in study guidelines, manuals, and plans rather than the protocol; leaving certain criteria up to the “discretion of the investigator”; and not classifying missing data due to subjects being withdrawn from the study as protocol deviations. 13 –15
CAPA Program
Companies involved in clinical trials should have CAPA programs with corrective and preventive measures in place to minimize the impact of deviations on subject safety and data integrity and to prevent the reoccurrence of similar or related deviations in the future. These programs must be reflected in the organizations’ quality systems and SOPs. All individuals involved in the trial must be trained and familiar with following procedures relevant to their activities.
Detection and Timely Reporting
Protocol deviations can be detected by various personnel involved in the conduct, data review, analysis, or reporting of clinical trials. Procedures that permit early detection and immediate corrective and preventive actions result in minimizing the impact on subject safety and integrity of trial data. Figure 1 displays the impact of the deviation (cost of quality) on the vertical axis versus the time point of detection in the life cycle of a clinical trial (time) on the horizontal axis.
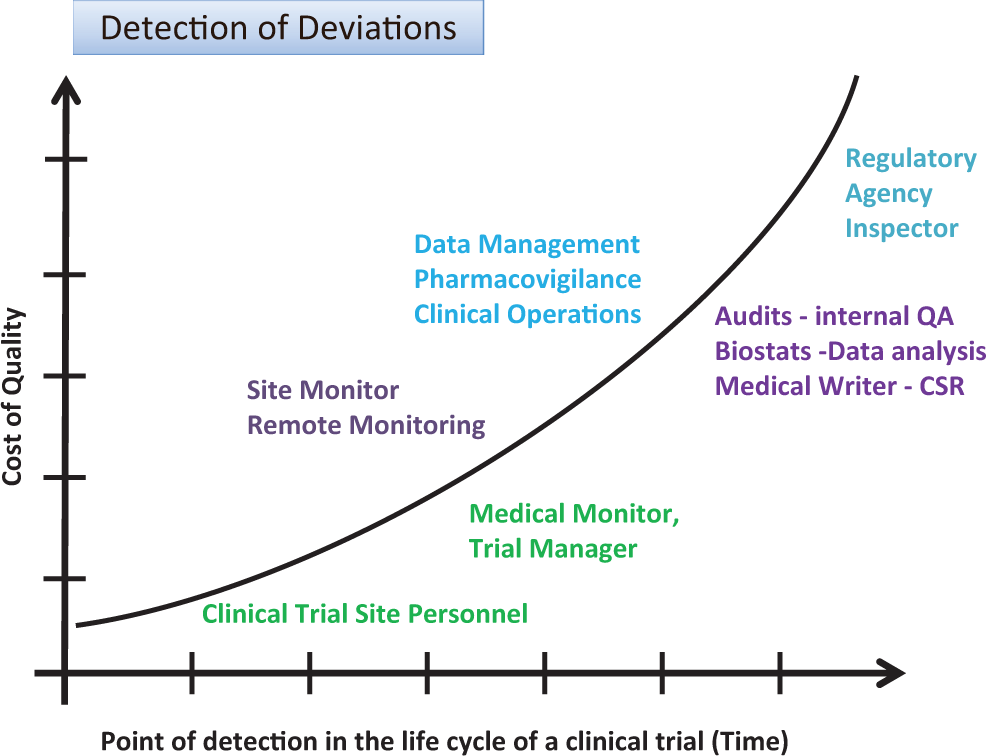
Detection of deviations: cost of quality and point of detection in time.
Table 1 summarizes the different functional groups who may be involved in the detection of protocol deviations during the conduct of a clinical trial and the process they should follow for discovering, reporting, and following up of such protocol deviations. Table 1 describes an approach that illustrates the various stages related to detection and reporting of protocol deviations in a clinical trial. Traditionally, these processes have been sequential, but current approaches with real-time capture of data in EDC systems and devices that directly capture data from subjects (eg, Holter monitors or subject diaries), as well as risk-based monitoring approaches (see “Guidance for Industry: Oversight of Clinical Investigations—A Risk-Based Approach to Monitoring” 16 and “Reflection Paper on Risk-based Quality Management in Clinical Trials 17 ), are changing who does what when as the study progresses and data are available. Thorough training of site personnel with frequent and open communications between sites and sponsor personnel can minimize the occurrence and impact of deviations. Current improvements in the conduct of clinical trials include sites directly uploading key study documents into electronic trial master files (eTMF) permitting remote review of these documents. Careful review of these can help identify protocol deviations at sites (eg, drug storage temperature excursions). In addition, use of signal detection methods across systems that contain study data or metadata can permit either prevention or early detection and follow-up.
Stages related to the detection and reporting of protocol deviations.
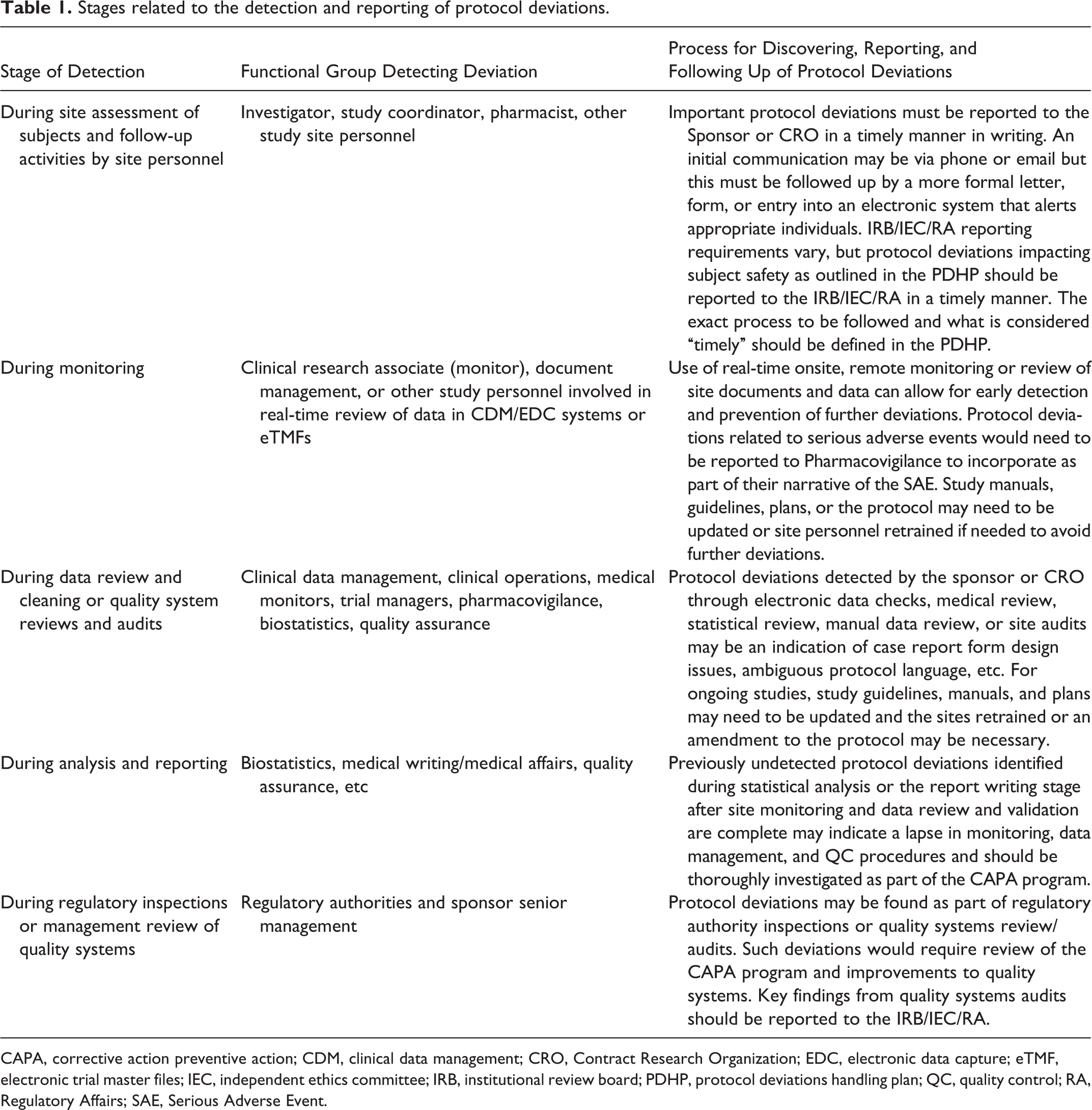
CAPA, corrective action preventive action; CDM, clinical data management; CRO, Contract Research Organization; EDC, electronic data capture; eTMF, electronic trial master files; IEC, independent ethics committee; IRB, institutional review board; PDHP, protocol deviations handling plan; QC, quality control; RA, Regulatory Affairs; SAE, Serious Adverse Event.
Clinical research associates (monitors) must be experienced and adequately trained on the protocol and provided procedures and tools to assist verification of adherence to the protocol as well as to regulatory and GCP requirements by site personnel. If onsite monitoring visits are planned, monitors must be provided adequate time at sites to verify the data and check regulatory documents during their monitoring visits, as well as communicate with and, if needed, retrain site personnel as well as identify site-specific possible root causes for deviations. All protocol deviations should be documented in accordance with SOPs and the PDHP and communicated to the investigational site.
As part of the data validation process, electronic checks are executed and data often manually reviewed by CDM personnel, pharmacovigilance, medical monitors, and biostatisticians. During this process, data clarification forms or data queries are generated for sites to resolve. Some of these checks relate to inclusion/exclusion criteria or confirm that study visits (or assessments) were performed in accordance with the protocol. Data values outside the acceptable range are either data errors or protocol deviations. For example, if a study is to enroll subjects between 18 and 65 years old, and a 68-year-old subject is enrolled into the study, a query would be generated for the site to verify that the enrolled subject was actually 68 years of age. The electronic check related to this out-of-range value will need to be “closed” as an out-of-range value and appear as such in the database. Such queries confirming discrepant values should be carefully reviewed to determine whether the data discrepancy is a data error or a protocol deviation and followed up accordingly as part of the CAPA program.
As described earlier, according to chapter 48 of the FDA’s “Compliance Program Guidance Manual, Program 7348.811,” 6 as well as the ICH E6 GCP guidance, sections 4.5.2, 4.5.3, and 4.5.4, 9 protocol deviations should be reported to the sponsor, to the IRBs or IECs (independent ethics committees), and, when required, to regulatory authorities in a timely manner. Additional local requirements for regulatory reporting might vary depending on the region.
In addition, the regulatory authorities (eg, FDA and European Medicines Agency [EMA]) expect that appropriate corrective and preventive actions are undertaken. CAPA systems are a requirement defined by ICH Q10 and are fairly common in manufacturing of medical products. As with other quality system elements, the development and execution of CAPAs are now being applied to the conduct of clinical trials. Once protocol deviations are identified, they should not only be classified (see Classification section) with respect to the impact to subjects’ safety and scientific integrity of study data but also evaluated for the underlying reason or “root cause” for the deviation (see Appendix 2 for additional details on CAPAs).
Tracking
It is important to detect and track all relevant protocol deviations as defined in the PDHP. Select protocol deviations, such as certain missing data or visits performed outside the permitted windows, can be extracted from the clinical database and summarized without requiring tracking in a separate system or a CAPA for each such individual missing item or out-of-visit assessment. However, these missing or out-of-window assessments should be evaluated in as close to real time as possible to ensure that they do not accumulate and affect subject safety or ability to draw meaningful conclusions from the data. Per the ICH E3 guidance, even though the CSR requires only reporting of important deviations, during inspections regulatory authorities may expect to be provided lists of all deviations. The sponsor PDHP or other study plans should address processes for review of protocol deviations electronically or in listings, as well as documentation of such reviews and appropriate corrective actions and preventive actions.
Once detected, protocol deviations should be documented and tracked, with corrective and preventive actions taken as appropriate. Ideally, protocol deviations should be tracked in a CTMS or a CDM/EDC system that complies with US Title 21 of the Code of Federal Regulations, Part 11, and/or ICH GCP, Section 5.5.3 (or other international applicable regulations or guidances). Such systems should include secure access, date and time stamps, audit trails, and so on. Based on the survey results (Appendix 1), roughly an equal proportion of companies use CDM/EDC systems, CTMSs, and spreadsheets and a small percentage are using quality risk digital dashboards to document or track deviations. While using spreadsheets provides ease of use, the lack of controls (eg, unauditable changes) may result in inadequate follow-up of potential protocol deviations or changes (intentional or unintentional) and lack of traceability. Protocol deviations should also be captured in monitoring reports or follow-up letters to sites after monitoring visits. While these are useful for documentation and follow-up, they do not provide a structured or systematic approach to track, classify, summarize, and follow up on protocol deviations.
CDM/EDC systems and CTMSs each provide their own advantages and disadvantages for tracking protocol deviations, and a detailed discussion of these is beyond the scope of this paper. Further work needs to be done to determine best practice as to which systems are more appropriate for tracking of protocol deviations, as well as their use and interoperability with enterprise quality management and other systems.
Classification
As part of risk assessment, it is important to be able to classify protocol deviations based on their impact on subject safety and data integrity for addressing the objectives of the study. SACHRP in its minutes of February 28-29, 2012, approved a recommendation on protocol deviations: This recommendation specifically addresses 3 types of deviations: Deviations that occur because an investigator, research staff or other party involved in the conduct of research intentionally decides to deviate from the approved protocol. Deviations from the protocol that are identified before they occur, but cannot be prevented. Deviations from the protocol that are discovered after they occur.
4
Deviations at the site may be intentional or unintentional, and the SACHRP minutes provide examples, classifying them as follows: Intentional deviations related to preventing a subject from harm Intentional deviations unrelated to preventing a subject from harm (eg, an investigator enrolls a subject who does not meet inclusions/exclusion criteria or may not be able to comply with all protocol requirements) Unintentional deviations that could not be prevented (eg, a subject calls and says that due to bad weather they cannot come in for their study assessment visit within the permitted window for that visit) Unintentional deviations discovered after the fact (eg, a subject did not take the study medication while at home)
4
The ICH E3 guidance for CSRs (see Reporting in the CSR section) suggests summarizing all important protocol deviations in the CSR and provides examples of what would be considered “Important.” Several companies now classify protocol deviations on the basis of these definitions only as “Important” or “Not Important,” whereas some still prefer using categories of “Critical,” “Major,” and “Minor.” While “Important” deviations are defined in the ICH E3 guidance, there is no consensus for definitions of Critical, Major, and Minor deviations. We propose the definitions below.
Critical Deviations
Those deviations that have a clear and direct negative effect on the rights, safety, or well-being of subjects or integrity of data for key conclusions related to efficacy or safety are considered critical deviations. Any fraud or misconduct, irrespective of whether a subject was harmed, is considered critical. Additional examples of critical deviations include enrolling patients that do not meet key eligibility criteria; incorrect administration of study drug; absence of source documents; and failure in recording or incorrectly recording the primary efficacy variable(s), serious adverse events, or other key assessments related to the therapeutic area and indication and potential safety concerns of the medical product. Critical protocol deviations should be reported to regulatory authorities and IRB/IECs, per country laws and policies, within defined timeframes.
Major Deviations
Those deviations that may have a reasonable negative effect on the rights, safety, or well-being of subjects or integrity of key data. Examples of major deviations include a pattern of deviations classified as minor or poor quality of the data. Other examples of major deviations include inaccurate recording, failure in recording, or incorrectly recording nonserious adverse events, secondary efficacy assessments, and laboratory safety data. Major protocol deviations should also be reported to regulatory authorities and IRB/IECs, per country laws and policies, within defined timeframes.
Minor Deviations
Those deviations that do not have an impact on the rights, safety, or well-being of the subjects or the integrity of key data. Examples of minor deviations may include not recording an individual item on the physical exam, the exact route or dosage of a concomitant medication that is not likely to affect safety, or other nonkey background, efficacy, or safety assessments.
These categorizations (Critical, Major, Minor, and Important, Not Important) can be complementary, and some organizations choose to use both. Figure 2 reinforces the relationship between Critical, Major, and Minor deviations and Important and Not Important protocol deviations. The former categorization helps in tracking and reporting, while the latter is primarily focused on summarizing in the CSR. Each approach offers its own advantages and disadvantages. Both classifications should be made early in the life cycle of a protocol deviation and can be updated as more information becomes available, with finalization by database lock. Some companies choose the 3-tiered approach to allow for a greater level of granularity to facilitate their internal processes for trending and tracking purposes and for reporting to IRBs/IECs and regulatory authorities. In addition, it is consistent with historical use and, at least for some sponsors, with their current QC and QA processes as well as with some of the risk management approaches currently implemented. The big disadvantage of the 3-tier approach is that each protocol deviation eventually still needs to be evaluated as Important or not for reporting in the CSR to be compliant with the ICH E3 guideline requirements. Organizations who use the 2-tiered approach feel it causes less confusion and simplifies their processes, as the categorization is consistent with ICH E3 reporting requirements from detection to reporting.
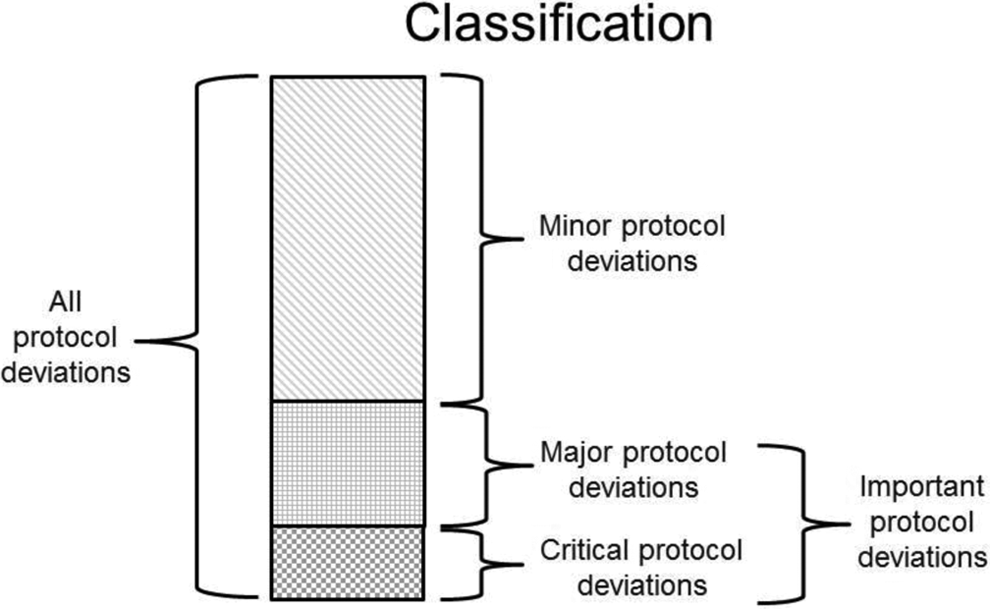
Categories of protocol deviations.
The results of the online survey as well as the workshop and presentations conducted as part of the preparation of this paper showed that there are strong individual preferences for both approaches. Since it is unlikely that industry will reach consensus in the near future, it is recommended that organizations prospectively define their classification approach in the PDHP.
Due to the wide variation in IRB/IEC and regulatory authority reporting requirements, neither of the above two classification schemes may offer a clear indication of what should be reported. While some companies may choose to report all Important or all Critical and Major deviations, others may choose to have systems in place to flag and identify which protocol deviations should be and were reported to IRB/ECs and regulatory authorities.
Permitting subjects who do not meet eligibility criteria at the investigational site to enroll into a clinical trial would, in general, be considered a Critical, Major or Important protocol deviation depending upon the eligibility criteria not met and its impact on subject safety or integrity of key data. However, such deviations should always be classified as Important, with respect to reportability in the CSR, in line with ICH E3. For example, including a subject who does not have a targeted disease may result not only in potential harm to the subject, but also confound the results with invalid data. In addition, including such a patient may also violate their rights and have consequences on insurance coverage.
It might be difficult to prospectively categorize all protocol deviations and assess their impact on data integrity. For example, one missing trial assessment might not be Major or Important, but repeated missed assessments might have an impact on data integrity or subject safety and indicate an overall lack of compliance (by the subject or site personnel). If such protocol deviations are not tracked and appropriately followed up, they might stay unnoticed. Hence, in addition to deviation categories defined in the PDHP, guidance should be provided as to the extent of repeated Minor or Not Important deviations that would be cause for greater concern.
Reporting in the CSR
Per the ICH E3 guidance on the structure and content of CSRs, section 10.2, All Important deviations related to study inclusion or exclusion criteria, conduct of the trial, patient managements or patient assessment should be described. In the body of the text, protocol deviations should be appropriately summarized by center and grouped into different categories, such as: Those who entered the study even though they did not satisfy the entry criteria. Those who developed withdrawal criteria during the study but were not withdrawn. Those who received the wrong treatment or incorrect dose. Those who received an excluded concomitant treatment. In Appendix 16.2.2, individual patients with these protocol deviations should be listed, broken down by center for multicenter studies.
10
Minor deviations may be listed but do not require separate discussion in the text. Certain protocol deviations, such as not recording a subject’s weight during a vital sign assessment or a missed question on a physical examination, may not need to be reported separately as an Important, Critical, or Major protocol deviation and may be obvious and implicit in the data displays as missing data. However, if the missing assessment is a key safety assessment (eg, weight in a study of subjects with unintended weight loss) then it should be highlighted in the data listings and analysis of protocol deviations (Listing 16.2.2 per ICH E3). Missing data can make interpretation of efficacy assessments difficult and as per the National Research Council recommendations that the FDA refers to, more emphasis should be paid to designing protocols and taking operational steps to minimize missing data. For more details the reader is referred to “The Prevention and Treatment of Missing Data in Clinical Trials” 18 and the EMA’s “Guideline on Missing Data in Confirmatory Clinical Trials.” 19
As per the ICH E3 guidance, individual patient data listing 16.2.2 should provide a list of all important protocol deviations by site.
Archival
As part of archival of a clinical trial, details of protocol deviations should also be archived. This would include systems where information related to protocol deviations were tracked and managed and would include data in CDM/EDC systems and CTMSs, the trial master file or eTMF, the CSR, and all its appendices.
Handling of Subjects Who Do Not Meet Study Eligibility Criteria
One of the most controversial issues during the conduct of clinical trials is whether subjects who “almost” meet eligibility criteria should be enrolled. The hard cutoff boundaries (eg, age younger than 65 years) can be somewhat arbitrary. With enrollment of patients into clinical trials being a constant challenge, some sponsors tend to allow latitude in subjects being enrolled. This section describes the management of subjects enrolled into a trial who do not meet the protocol entry criteria or are retrospectively determined to not meet one or more of the eligibility criteria.
Enrollment of subjects not meeting eligibility criteria may jeopardize subject safety and welfare and may result in questions concerning the validity and integrity of the data collected. Many sponsors feel that granting a prospective “waiver” somehow legitimizes the protocol deviation. From the perspective of GCP as well as analysis and reporting of data, the presence or absence of a waiver does not make any difference regarding subject safety or data integrity. A waiver only indicates that the sponsor was aware of the deviation and agreed to it prospectively. Most sponsors have moved to “zero tolerance” of such deviations and do not permit subjects who do not meet eligibility criteria to be enrolled into a study. We support and strongly recommend this approach. The GCP Q&A section on the EMA website states the following about protocol waivers and their impact on subject safety and the interpretation of study results: Sponsors and investigators should not use systems of prospectively approving protocol deviations, in order to effectively widen the scope of a protocol. Protocol design should be appropriate to the populations required and if the protocol design is defective, the protocol should be amended. GCP does permit deviations from the protocol when necessary to eliminate immediate hazards to the subjects but this should not normally arise in the context of inclusion/exclusion criteria, since the subject is not yet fully included in the trial at that point in the process. GCP inspectors have observed a number of sponsors implementing systems where the investigator can contact the sponsor, usually the Medical Monitor, and request a prospective approval to deviate from the inclusion and/or exclusion criteria. The use of such systematic waiver systems in clinical trials is not considered to be appropriate and studies using such a system might be regarded as non-compliant with GCP.
20
Overall Conclusions and Best Practices
We concur with SACHRP’s central recommendation that “FDA and OHRP publish a clear statement of their positions regarding intentional protocol deviations.” 4
Due to the growing complexity and nature of clinical trials and since clinical trials require subjects to willingly participate in them, it is unrealistic to expect to completely avoid protocol deviations. However, by considering the following, the occurrence and impact of protocol deviations, especially Critical and Major ones, can be minimized: Developing a well-designed and carefully reviewed clinical trial protocol with adequate input and consideration from multiple investigators, study coordinators, and site personnel who see patients on a day-to-day basis. Standardizing terminology related to protocol deviations. Having systems and procedures and following them to ensure adequate oversight and controls of individuals involved in the trial whose work affects the life cycle of protocol deviations. Implementing a CAPA program with SOPs. Understanding the life cycle of protocol deviations and developing best practices for classifying and managing deviations in PDHPs. Developing SOPs that describe the process, roles, and responsibilities for handling protocol deviations through their entire life cycle from prevention through detection, tracking, classification, reporting, and archival. Conducting effective and regular training on the protocol, study manuals, guidelines, and plans for all individuals involved in the trial. Training should be conducted prior to the first subject being enrolled into the study at the investigator meeting or at site initiation monitoring visits. Real-time remote or on-site monitoring of study data and documents through CDM/EDC, CTMS, and eTMF systems to confirm adherence to the protocol, GCP, and regulatory requirements. Using risk management dashboards and signal detection tools to more closely approach real-time review and monitoring of key study data and as well as protocol deviations. Routine review and reconciliation of the clinical database, monitoring reports, and other sources, tracking protocol deviations for timely detection, trend analysis, and follow-up of unreported important, critical, and major deviations. Classification of protocol deviations based on criteria prespecified in the PDHP and finalization of classification prior to database lock and study unblinding. Timely implementation of corrective actions and preventive actions. Appropriate and timely reporting of protocol deviations (eg, to sponsors, IRB/IEC, and, if necessary, regulatory authorities). Proper and complete handling in statistical analyses and reporting of important protocol deviations in the CSR. This may include select analyses for a per-protocol data set in addition to an efficacy evaluable data set. Instituting ongoing quality improvement systems and lessons learned with respect to handling of protocol deviations.
As the DIA Good Clinical Practice and Quality Assurance community continues to explore the topic of protocol deviations, future directions will encompass discussions of the best practices presented above including development of a template PDHP and SOPs.
Footnotes
Acknowledgments
The authors would like to thank Leslie M. Sam, Director, Safety, Efficacy, Customer Information and Global Product Compliants, Global Quality Systems, ELi Lilly and Company, who was Chair of the GCP and QA Community during the development of this publication and led the team effort for writing this paper. In addition, the authors acknowledge the following individuals: Faiz Ahmad, statistician, GSK; Anne Blanchard, RAB, Blanchard & Asociados/Blanchard & Associates; Scott Brand, PhD, director, strategic planning and quality assurance, KAI Research; Bonnie Corrigan, Corrigan Medical Writing; Sylvia Engelen, Bayer HealthCare Pharmaceuticals Inc; Vicky Foster, GSK; Susan Kenna Franks, Veritas Clinical Consultants; Heather Hill, The EMMES Corp; Noelle Holten Pind, NovoNordisk; Brent Ibata, PhD, JD, MPH, RAC, director of operations, Sentara Cardiovascular Research Institute; Jamila Joseph, FCMI, head, clinical research, Reliance Life Sciences Pvt Ltd; Susy Laws, SLD Solutions; Felix Khin-Maung-Gyi, PhD, MBA, CIP RAC, CEO, Chesapeake IRB; Estela Lamarca, associate director–global development quality, Bayer; Laurin Mancour, program director/instructor, Durham Technical Community College, CT Research Associate Program; Wendy Mangels, Vertex Pharmaceuticals; Brian Maurer, director, clinical development and medical affairs, Forest Research Institute Inc; Margaret Newman, report writer/editor, CompleWare Corp; Linda Nichols, Sunstone Medical Research; Robert Palmer, senior director, Forest Research Institute Inc; Nancy Perrella, JD, RAC, regulatory policy & compliance, Covance Inc; Neil Sankar, physician, clinical development consultant; Yukiko Savini, Savinin Consulting (data management); Doris Schweighofer, country clinical research manager, Bayer HealthCare Austria; Michaela Sommer, German Interdisciplinary Sarcoma Group (GISG); Elana Stolpner-Miller, MD, physician; Uschi Stoutenburgh, director of clinical operations, Cubist Pharmaceuticals; John Weiler, president, CompleWare Corp; Beat Widler, PhD, independent CQA & quality risk management expert, managing partner at Widler & Schiemann AG; and Andrea Wohlsen, PhD, BfArM.
Author Note
Presented at the DIA 25th Annual EuroMeeting, March 5, 2013, Amsterdam, The Netherlands; DIA 49th Annual Meeting, June 27, 2013, Boston, MA, USA; DIA 7th Annual Clinical Forum, October 8-9, 2013, Dublin, Ireland; and DIA China, Root Cause Investigations and Corrective Actions for GCP Compliance, September 2-3, 2013, Chengdu, China.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.