
Abstract
Background:
Pediatric safety studies are conducted for drugs undergoing development for use in pediatric patients. The objective of this study was to describe safety studies and compare adverse events of antiretroviral drugs between pediatric patients and adult subjects.
Methods:
Pediatric and adult adverse event data were obtained from US Food and Drug Administration (FDA)–approved drug labels for 9 antiretroviral drugs with pediatric indications approved by the FDA prior to 2013. For adverse events (AEs) reported in both pediatric patients and adult subjects, the risk difference (RD) and associated confidence interval (CI) were calculated.
Results:
Of 35 drug-AE combinations, 10 AEs were reported at statistically significantly (P < .05) higher incidence rates in the pediatric population than in the adult population, and 3 AEs were reported at statistically significantly higher rates in the adult population than in the pediatric population. The largest differences where the risk of an AE was greater in pediatric patients than in adult subjects were for rash with efavirenz (RD = 36.24% [95% CI, 21.1 to 50.53]), diarrhea with efavirenz (RD = 24.53% [95% CI, 9.06 to 39.57]), and rash with nevirapine (RD = 16.14% [95% CI, 9.7 to 24.27]). The largest differences where the risk of an adverse event was lower in pediatric patients than in adult subjects were for headache with abacavir (RD = –12% [95% CI, –16.81 to –6.89]), diarrhea with tipranavir (RD = –11.32% [95% CI, –15.02 to –5.6]), and diarrhea with lamivudine (RD = –9.88% [95% CI, –15.91 to –3.98]).
Conclusions:
The adult adverse event experience provides preliminary data for pediatric drug safety, yet the specific types of adverse effects and frequencies may not be predicted in children based exclusively on adults. As adult safety data do not fully inform the pediatric safety profile, pediatric safety studies should continue to be conducted separately for drugs undergoing testing in pediatric patients.
Introduction
Legislation and regulation in the United States and Europe has stimulated pediatric drug development over the past 2 decades. The strategy for a pediatric drug development program is dependent on assumptions regarding disease progression and response to intervention in children compared with adults. In some cases, extrapolation of efficacy can be applied, which is an approach to increase the efficiency of pediatric drug development programs and allow pediatric patients timely access to safe and effective medications. 1 The ability to extrapolate effectiveness is dependent on assumptions regarding the similarity in the course of the disease, the effects of the drug in adult subjects and pediatric patients, and exposure-response relationship between adult subjects and pediatric patients. While efficacy may be extrapolated in some cases, the safety profile of drugs in children may not be extrapolated because adults are not developing and undergoing all of the neurologic, endocrine, metabolic, and orthopedic changes that occur in the pediatric population. Therefore, for a product that is to be studied for use in the pediatric population, safety studies are required in pediatric patients.
Therapy for human immunodeficiency virus (HIV) infection is one therapeutic area in which pediatric drug development has advanced considerably. Clinical trials of antiretroviral (ARV) drugs in HIV-infected children have improved the pharmacological management of pediatric HIV, resulting in a dramatic reduction in morbidity and mortality. 2,3 Antiretroviral drugs are routinely recommended for evaluation in the pediatric population. To date, approximately 25 ARV drugs and combination products are approved for use in the pediatric population. 4 The effectiveness of ARV drugs for HIV infection can be extrapolated from adequate and well-controlled studies in adults when supplemented with safety and pharmacokinetic-pharmacodynamic (PK-PD) studies conducted in children.
Comparisons between adult and pediatric safety studies may provide insight into study design issues that affect the value of the information obtained in a pediatric safety study. The objective of this study was to describe safety studies and compare adverse events of ARV drugs between pediatric patients and adult subjects as contained in Food and Drug Administration (FDA)–approved drug labels.
Methods
Clinical trial reports of adverse events (AEs) were reviewed in the labels of noncombination ARV drugs with pediatric indications approved by the FDA prior to 2013. Drug labels that provided the incidence of both pediatric- and adult-specific AEs were identified. Labels included quantitative AE data and used a frequency threshold whereby the observed incidence rate of an AE was reported only if it was equal to or greater than the threshold value. Standardized definitions of AEs were taken from the Common Terminology Criteria for Adverse Events (CTCAE) v4.0.
5
For AEs that were reported in both pediatric patients and adult subjects, the risk difference (RD) of the AE was calculated as

where risk is defined as the incidence of the AE in FDA labeling. Risk differences are presented as a percentage and were chosen in part because they are always defined and employ an absolute scale, whereas other metrics using a relative scale, such as odds ratios or risk ratios, may be undefined under some circumstances. The 95% confidence interval (95% CI) of the RD was calculated using Miettinen-Nurminen score intervals, 6,7 as implemented in the R 8 package PropCIs. 9 Miettinen-Nurminen score intervals have better statistical coverage properties than do traditional Wald intervals. 10
Adverse events that were reported in pediatric patients but below the frequency threshold in adult subjects were recorded. Likewise, AEs that were reported in adult subjects but below the frequency threshold in pediatric patients were also recorded. Statistical comparisons of these data were not performed, as the actual incidence rate in 1 population was unknown (ie, below the frequency threshold but not necessarily zero).
Results
A total of 17 unique noncombination ARV drug products were identified, of which 9 contained the incidence of both pediatric- and adult-specific AEs. The characteristics of the pediatric and adult safety studies for these products, including background therapy, are presented in Table 1. On average, the pediatric safety studies included 129 ± 64 patients, and the adult safety studies included 437 ± 318 subjects. The lower bound of the age range included in the pediatric safety studies ranged from 6 weeks to 6 years, while the upper bound ranged from 14 to 17 years.
Study Characteristics.
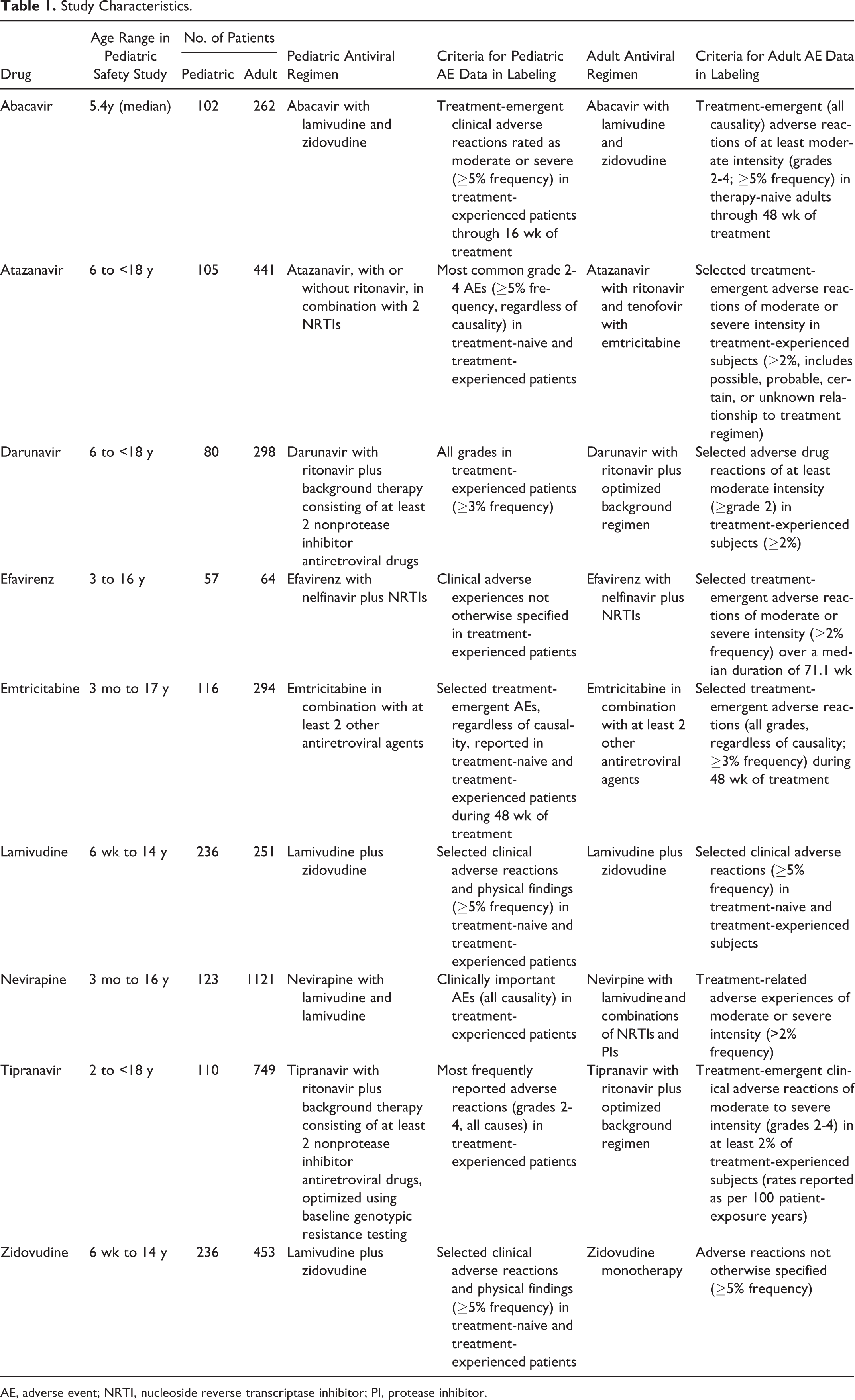
AE, adverse event; NRTI, nucleoside reverse transcriptase inhibitor; PI, protease inhibitor.
The source and criteria for inclusion of AE information for adult subjects and pediatric patients in FDA-approved labels often differed. The criteria for the inclusion of AE information in labeling were different between drugs as well as between pediatric patients and adult subjects for the same drug. Adult AEs reported in labeling were generally selected adverse reactions deemed related to the study medication by investigator assessment. Pediatric AEs were more often reported for all causality, regardless of relationship to the study medication.
The incidence of some AEs was significantly different between adult subjects and pediatric patients. For the protease inhibitor class, AEs that were significantly more likely to be detected with atazanavir treatment in pediatric patients compared with adult subjects included diarrhea (RD = 6.53% [95% CI, 2.2 to 13.55]), rash (RD = 11.34% [95% CI, 5.59 to 19.42]), and jaundice (RD = 10.25% [95% CI, 4.13 to 18.55]) (Figure 1). These findings were all statistically significant, since the 95% CIs do not include zero, although it is unclear if these differences are clinically meaningful. The background therapy in pediatric patients included 2 nucleoside reverse transcriptase inhibitors (NRTIs) with or without ritonavir. The background therapy in adult subjects included ritonavir, tenofovir, and emtricitabine (Table 1). For darunavir (in combination with ritonavir), vomiting was significantly more common in pediatric patients (RD = 7.84% [95% CI, 1.83 to 16.05]). Conversely, pediatric patients were less likely to experience diarrhea with tipranavir (in combination with ritonavir) (RD = –11.32% [95% CI, –15.02 to –5.6]). Additionally, several AEs were reported in pediatric patients but were below the reporting frequency threshold in adults, including cough and fever with atazanavir, which were reported in 21% and 18% of pediatric patients, respectively (Table 2).
AEs Reported in Pediatric Patients but Below the Frequency Threshold in Adults.a

AE, adverse event; NR, not reported.
aNo adverse events fit the criteria for darunavir.
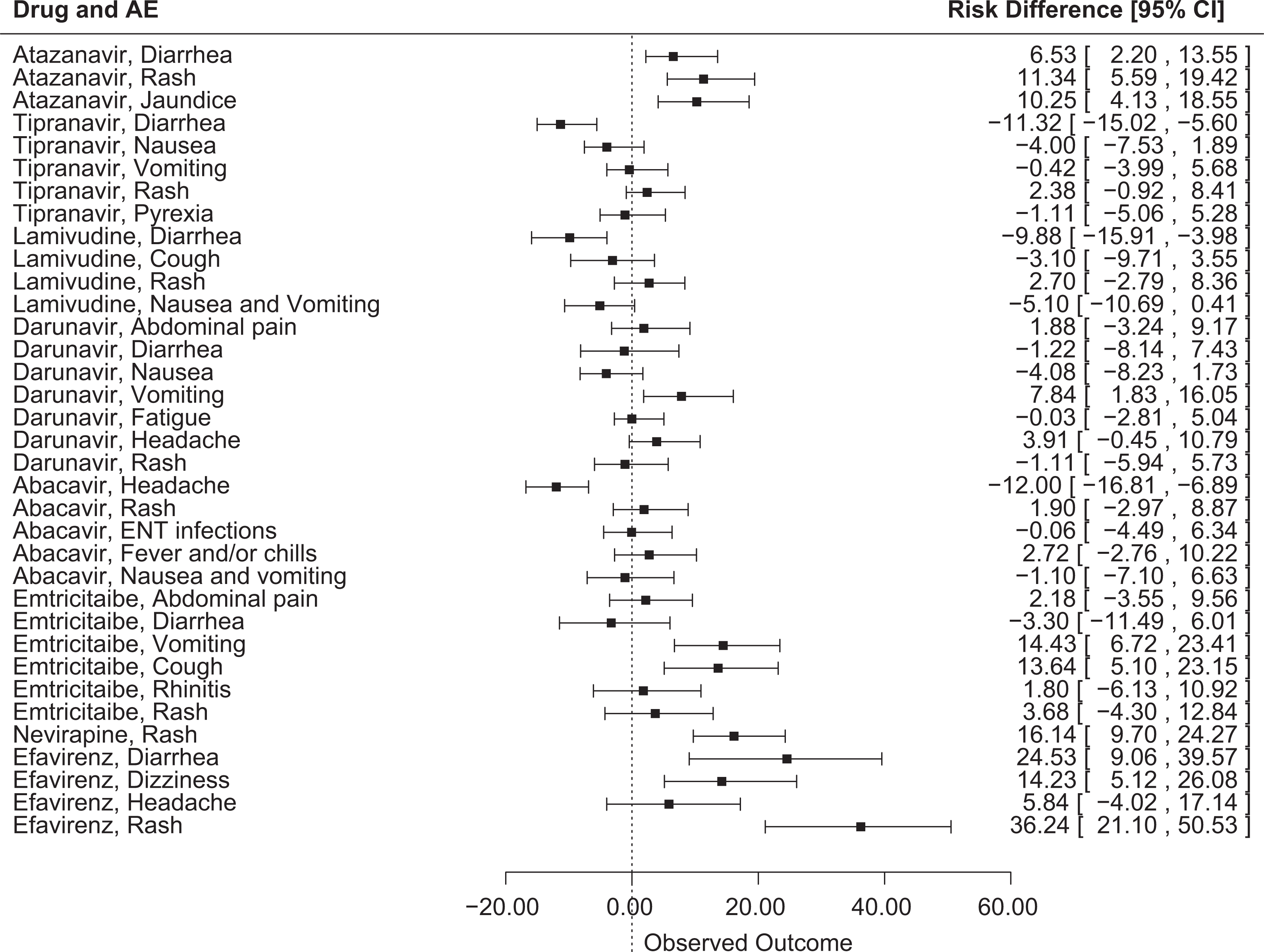
Risk difference and 95% confidence interval for adverse events in pediatric patients versus adult patients for antiretroviral drugs.
For the NRTI class, vomiting and cough were significantly more likely to occur in pediatric patients in emtricitabine safety studies (RD = 14.43% [95% CI, 6.72 to 23.41] and RD = 13.64% [95% CI, 5.1 to 23.15], respectively) (Figure 1). Headache was less frequently reported in pediatric patients receiving abacavir (RD = –12% [95% CI, –16.81 to –6.89]). Fever was reported in 25% of pediatric patients in the zidovudine and lamivudine pediatric safety studies but was not above the reporting threshold (5%) in adult subjects. In addition, in pediatric patients receiving emtricitabine, infection was reported in 44% of patients and skin hyperpigmentation was reported in 32% of patients, although these AEs were not above the reporting threshold in adult subjects (Table 2). Adverse events that were reported in adult subjects but below the reporting frequency threshold in pediatric patients included headache (63% for zidovudine and 35% for lamivudine) and nausea (51% for zidovudine, 33% for lamivudine, 19% for abacavir, and 18% for emtricitabine) (Table 3).
AEs Reported in Adult Patients but Below the Frequency Threshold in Pediatric Patients.

AE, adverse event; NR, not reported.
For the nonnucleoside reverse transcriptase inhibitor (NNRTI) class, rash was significantly more likely to occur in pediatric patients receiving nevirapine compared with adult subjects (RD = 16.14% [95% CI, 9.7 to 24.27]) (Figure 1). In the efavirenz safety studies, AEs that were significantly more common in pediatric patients compared with adult subjects included diarrhea (RD = 24.53% [95% CI, 9.06 to 39.57]), dizziness (RD = 14.23% [95% CI, 5.12 to 26.08]), and rash (RD = 36.24% [95% CI, 21.1 to 50.53]). The reporting threshold affected comparison in some cases: in efavirenz safety studies, fever was reported in 21% of pediatric patients but was not above the reporting threshold in adult subjects; 13% of adults experienced pain, which did not exceed the reporting threshold in pediatric patients (Tables 2 and 3).
Discussion
Evaluation of drug safety is an essential component of drug development programs. Safety studies allow investigators and drug regulators to detect serious safety concerns and to identify and estimate the frequency of common nonserious adverse events. A thorough evaluation of adverse events allows patients and clinicians to make informed treatment decisions and to anticipate, and perhaps mitigate, adverse events that may be causally related to the use of the drug. In the treatment of HIV infection, characterization of drug safety is particularly important since it is a major distinguishing factor between antiretroviral regimens. 11
In the setting of pediatric drug development, pediatric safety studies are always requested by the FDA. Because of developmental differences, some pediatric adverse events may not be identified, or their incidence not captured, in adult studies. 12 Human immunodeficiency virus is a therapeutic area that allows for a comparison of adverse event incidence between pediatric patients and adult subjects due to the large number of pediatric trials that have been conducted. The exposure-efficacy response relationship for HIV drug products is not expected to change with age, which creates a scenario whereby dosing regimens in pediatric patients are designed to target drug exposures shown to be efficacious in adult subjects. Therefore, comparisons of adverse events are in situations where drug exposure is similar, and differences in adverse events between adult subjects and pediatric patients would not be attributed to differences in dosing or drug concentrations.
Across the ARV drug classes, this study considered a total of 35 drug-AE combinations. Of these, 10 were reported at statistically significantly (P < .05) higher incidence rates in the pediatric populations than in the adult population, and 3 were reported at statistically significantly higher rates in the adult population than in the pediatric population. Taken together, these results provide evidence that pediatric and adult drug safety are different with respect to adverse event frequencies and, in some cases, specific types of adverse events. The results of a safety study in one population can be viewed as no more than suggestive when considering the risk profile faced by the other population.
Improving FDA labeling with new pediatric information has been an important outcome of the passage of the Best Pharmaceuticals for Children Act (1997) and the Pediatric Research Equity Act (2003). Over 450 label changes are the result of new pediatric studies, 13 and many of these changes relate to drug safety in pediatric patients. One of the improvements that will occur over time relates to the manner in which this information is presented in drug labels. The Physician Labeling Rule (PLR), initiated by the FDA in 2006, promotes the standardization of safety in labeling. 14 The PLR requires adverse reactions in labeling include (1) a listing of the most frequently occurring adverse events and (2) the criteria used to determine inclusion (eg, frequency cutoff rates). 15 This study found that the criteria for the inclusion of adverse event information often lacked standardization between drugs as well as between pediatric patients and adult subjects for the same drug. For example, for darunavir, all grades of adverse reactions were reported for pediatric patients while only selected adverse drug reactions of at least moderate intensity (≥grade 2) for adult subjects were reported. For this reason, comparisons may be subject to bias. Further standardization of safety information for drugs within a therapeutic category may make this information more useful.
Previous studies have shown that AEs in children differ from those in adults across a broad array of drugs in therapeutic areas outside of HIV. For example, neuropsychiatric adverse events in children that were either unexpected or greater in frequency than anticipated based on adult data were identified for 12 products spanning multiple therapeutic areas that were reviewed by the FDA between 1997 and 2007. 16 The unanticipated incidence of neuropsychiatric adverse events in children highlights the need for safety trials in the pediatric population. In addition to premarket safety evaluations, postmarketing assessments are also necessary since many safety signals are not captured during the development process. Current legislation in the United States requires the FDA to report to its Pediatric Advisory Committee on adverse events occurring during the 18-month period after granting pediatric exclusivity. A 2008 report found that the Pediatric Advisory Committee made specific recommendations for labeling changes for 17.9% of drugs reviewed between June 2003 and April 2007. 17 Therefore, the full scope of drug safety is often not captured prior to approval.
Several factors may contribute toward the observed differences in the incidence of adverse events. First, the developmental age of pediatric patients may complicate adverse event reporting. This is especially true of preverbal patients in whom some adverse events, such as central nervous system effects, are difficult to capture. Other events such as headache and nausea are unlikely to be communicated by younger pediatric patients. Next, in most cases, the adult safety profile was known prior to initiation of the pediatric trials, which may have increased reporting for adverse events that were expected. Additionally, adult safety studies included larger sample sizes than their pediatric counterparts. On average, adult studies included 3.67 times more subjects than pediatric studies and typically included a comparator group that aided in assessing the safety profile. Current 2013 FDA Guidance for Industry for the development of antiretroviral drugs recommends different sample sizes for adult and pediatric studies. For treatment-naive adults or adults with limited prior treatment experience, at least 500 subjects should be studied for 48 weeks. For heavily treatment experienced-adult subjects, 300 to 500 subjects studied for 24 weeks is recommended. In contrast, a safety database with at least 100 pediatric patients treated for 6 months is recommended due to the feasibility of enrolling in a pediatric clinical trial given that fewer patients are available to study. 18 This sampling is in line with safety studies in rare disease drug development programs at the FDA. 19
Due to exposures in environments like daycare centers and school, as well as developing immune systems, pediatric patients may experience more illnesses unrelated to HIV infection. As a result, background symptoms such as cough, fever, and gastrointestinal upset are likely to be more common. Without a safety comparator group, it is difficult to ascertain if such symptoms are true adverse events causally related to the study medication. Also, for events such as nausea and vomiting, formulation may have an impact on AEs. Liquid formulations with poor palatability may induce more nausea and vomiting in pediatric subjects than a tablet formulation in adults. Nonetheless, in safety studies for darunavir, vomiting was significantly more common in pediatric patients than in adult subjects, with both groups receiving an adult tablet formulation.
In this study, differences in adverse event frequency reported in FDA-approved labels were evaluated. Rare adverse events may also occur with antiretroviral drugs. The detection of rare adverse events in pediatric patients in the premarketing phase is further complicated by the small sample sizes included in safety studies. In order to have 95% certainty of detecting at least 1 case of an adverse event that is found in between 1 and 6 patients per 1000, a sample size of between 500 and 3000 patients is necessary. 20 As discussed at a 2010 FDA Pediatric Safety Surveillance Workshop, 21 premarketing clinical trials usually cannot address all safety issues reliably, and new methods of postmarketing surveillance for pediatric adverse events are necessary. While postmarketing surveillance using spontaneous reporting fills a gap in the detection of rare adverse events, previous studies have illustrated that serious adverse events in pediatric patients may be underreported in the postmarketing setting. Moore et al 22 showed that after adjusting for medication use and the size of the population, the number of serious adverse events reported to the FDA for pediatric patients between 1998 and 2005 was only 54% of expected. The reasons for this finding are unknown, but it may suggest the existence of barriers for reporting rare pediatric AEs.
The current study has limitations. Clinical trials are conducted under varying conditions, and the rates of AEs between trials may not directly comparable. In clinical trials for HIV infection, background therapy may differ between studies, making it difficult to attribute a specific adverse event to one particular drug. Additionally, follow-up duration may differ between pediatric and adult drug safety studies, and the relevance of the observed differences may vary in the clinical setting. The frequency of adverse events appearing in FDA-approved drug labeling was used for this study, and safety data included in labeling may be all-causality or only those AEs that are deemed as having a possible, probable, certain, or unknown relationship to the drug. This information is not always explicitly clear in drug labeling, nor is it necessarily the same for adult and pediatric adverse reactions from clinical trials within the same label. Finally, for abacavir, treatment-naive adult subjects were used for adverse event labeling while pediatric treatment-experienced patients were used. While many ARVs have different dosing regimens for treatment-naive versus treatment-experienced subjects, the dosing regimens used in the clinical trials to characterize drug safety (abacavir 300 mg twice daily for adults; abacavir 8 mg/kg twice daily for pediatric patients) yield similar drug exposure.
In summary, advances have been made in improving pediatric drug safety through labeling and drug warnings. Since safety cannot be extrapolated from adults, pediatric safety studies and postmarketing surveillance are necessary to capture the anticipated adverse event profile of a drug in children. This information allows clinicians to make rational drug selection and dosing decisions and is relevant for ARVs where toxicity is often a major differentiating factor between regimens. The collection of pediatric safety data is evolving as consortia are forming that use electronic medical records for pediatric patients. In addition, new methods in analyzing postmarketing surveillance data may serve to identify pediatric adverse events that were not captured during drug development due to sample size limitations. A better understanding and interpretation of pediatric safety studies is needed for optimal pediatric drug development and labeling. Although the adult adverse event experience provides preliminary data for pediatric drug safety, the specific types of adverse effects and frequencies may not be predicted in the pediatric population based solely on the adult experience. Therefore, pediatric safety studies should continue to be conducted separately for drugs undergoing testing in pediatric patients.
Footnotes
Acknowledgments
The authors thank Linda Lewis and Dianne Murphy for their constructive suggestions regarding this manuscript.
Author Note
The opinions expressed in this work are those of the authors and should not be interpreted as the position of the US Food and Drug Administration.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.