
Abstract
The field of oligonucleotide (OGN)–based therapeutics has been growing dramatically in the past decade, providing innovative platforms to develop agents for the treatment of a wide variety of clinical conditions. OGN agents have unique physicochemical properties and pharmacokinetic/pharmacodynamic characteristics. This review considers findings from the literature and information on new molecular entities submitted to the US Food and Drug Administration as OGN-based therapeutics. In addition, the article discusses several challenging issues from the perspective of clinical pharmacology, emphasizing the potential of immunogenicity, the effect of renal impairment on OGN exposure, drug-drug interactions, and the utility of pharmacokinetic/pharmacodynamic modeling. The field of OGN-based therapeutics is in evolution and will benefit from further studies as well as clinical experience to formulate guidelines and promote the development of this class of agents.
Keywords
Introduction
Oligonucleotide (OGN)–based therapeutics are a diverse group of nucleic acid–based drug products that utilize innovative molecular strategies to create novel treatments for a variety of medical conditions. These products include antisense OGNs (ASOs), RNA interference (siRNA), aptamers, immunomodulatory OGNs, and ribozymes. 1,2 Unlike conventional drugs, OGN-based agents in general are designed to act inside the cell, thus presenting unique challenges in drug design as well as issues in pharmacokinetics (PK) and pharmacodynamics (PD).
While these compounds are all based on nucleic acids, they can involve chemical modifications to promote target interaction, stability, and efficacy when acting intracellularly. Chemical modifications can also lead to PK and PD advantage. One example of such a modification in an ASO is the substitution of a sulfur moiety for a nonbridging oxygen moiety in the phosphodiester bond. This modification creates the phosphorothioate backbone and can lead to resistance of nuclease digestion. Additionally, OGNs can be modified at the 2′ position of the ribose sugar, such as 2′-O-methyl and 2′-O-methoxy-ethyl to increase antisense activity. In the case of ASOs, these modifications have been characterized in terms of their generation.
Through 2012, 121 unique new molecular entities for OGN-based therapeutics have been submitted as Investigational New Drug submissions to the US Food and Drug Administration (FDA). 3 Of these, more than half of the OGN Investigational New Drug applications are ASOs (Figure 1A), and many of these agents are in active development in phase 1 clinical trials (Figure 1B). The development of OGN-based therapeutics has expanded rapidly in the last decade (Figure 1C), with the majority of these agents intended for the treatment of cancer (∼50%), infection, cardiovascular diseases, inflammation, and diabetes (Figure 1D).
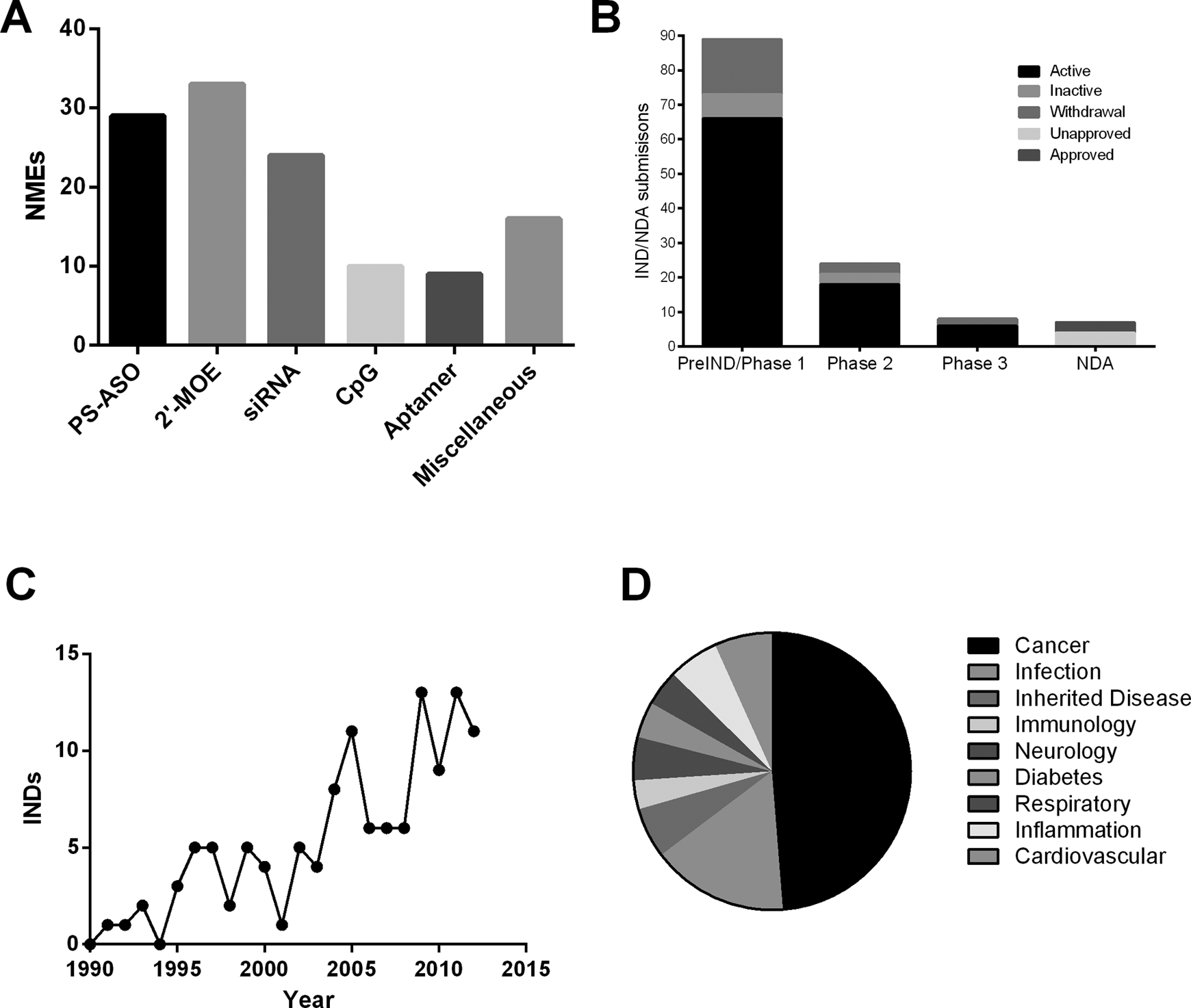
New Molecular Entity (NME) submissions to the US FDA from 1991 to 2012: (A) Investigational New Drug (IND) submissions by different classes of oligonucleotides, (B) IND/NDA submission by each phase of clinical development, (C) the original IND submissions by year, (D) IND submissions to the US FDA by therapeutic areas. INDs with the same molecular entity are counted as 1. The different therapeutic areas are displayed in a clockwise direction. PS-ASO, phosphorothioate antisense oligonucleotides without 2′-O-modification. 2′-MOE, 2′-O-methoxyethyl phosphorothioate oligonucleotides; CpG, immunomodulatory CpG oligonucleotides.
Thus far, 3 OGN products—Vitravene (fomivirsen, phosphorothioate ASO), Macugen (pegaptanib, an aptamer), and Kynamro (mipomersen, second-generation ASO)—have obtained approval by the FDA under new drug applications in 1998, 2004, and 2012, respectively. 2 Two of the products have antisense action, while pegaptanib is an aptamer. Fomiversin was approved for the treatment of cytomegalovirus retinitis, while mipomersen lowers cholesterol by its effects on apolipoprotein B. Pegaptanib has antiangiogenic activity for the treatment of age-related macular degeneration. While the following discussion can pertain to OGNs of all kinds, it uses ASOs for many examples because of extensive progress for that class.
Clinical PK
Any consideration of clinical PK should recognize the unique properties of ASO compounds and their fundamental differences from conventional small molecules, on the one hand, and biological agents, on the other. Thus, OGNs are larger than most small molecule drugs and are intended to act inside the cell. Indeed, the necessity for intracellular delivery in an adequate concentration is one of the defining features of these agents. Furthermore, ASOs target a nucleic acid and not a protein, imposing important structural requirements for target recognition.
In refining ASO for in vivo use, chemical modifications of OGNs can cause marked differences in PK profiles. 4 Among these modifications, the phosphorothioate backbone enhances hydrophobicity and protein binding as well as nuclease resistance. For the second-generation ASOs, the 2′-O-methoxy-ethyl modifications at the 5′ and 3′ termini limit susceptibility to the exonuclease cleavage. As a result, second-generation ASOs are primarily cleared by slow endonuclease digestion and ultimately by excretion of parent drug and metabolites in the urine. While the terminal elimination half-lives for the first-generation ASOs are typically <40 hours, the second-generation ASOs have longer half-lives, 14 to 30 days, allowing less frequent dosing. 5
In clinical trials, the efficacy and toxicity of ASOs are often dose dependent, and optimization of the dosing schedules may improve clinical outcomes. The first-generation ASOs often have short terminal half-lives, which may require a more frequent dosing schedule to attain the targeted drug levels. In oncology, for example, many of the first-generation ASOs have been administered as continuous prolonged (14-21 days) intravenous infusion to attain sustained pharmacologically effective plasma concentrations (Table 1). In contrast, the second-generation ASOs have much longer half-lives, allowing less frequent dosing. As these considerations suggest, prolonged dosing periods may be required to achieve steady state. Thus, when the clinical situation requires a rapid steady state or therapeutic effect, a loading dose regimen has been considered (Table 1).
Selected Examples of ASOs and the Dose Regimen in Clinical Trials.
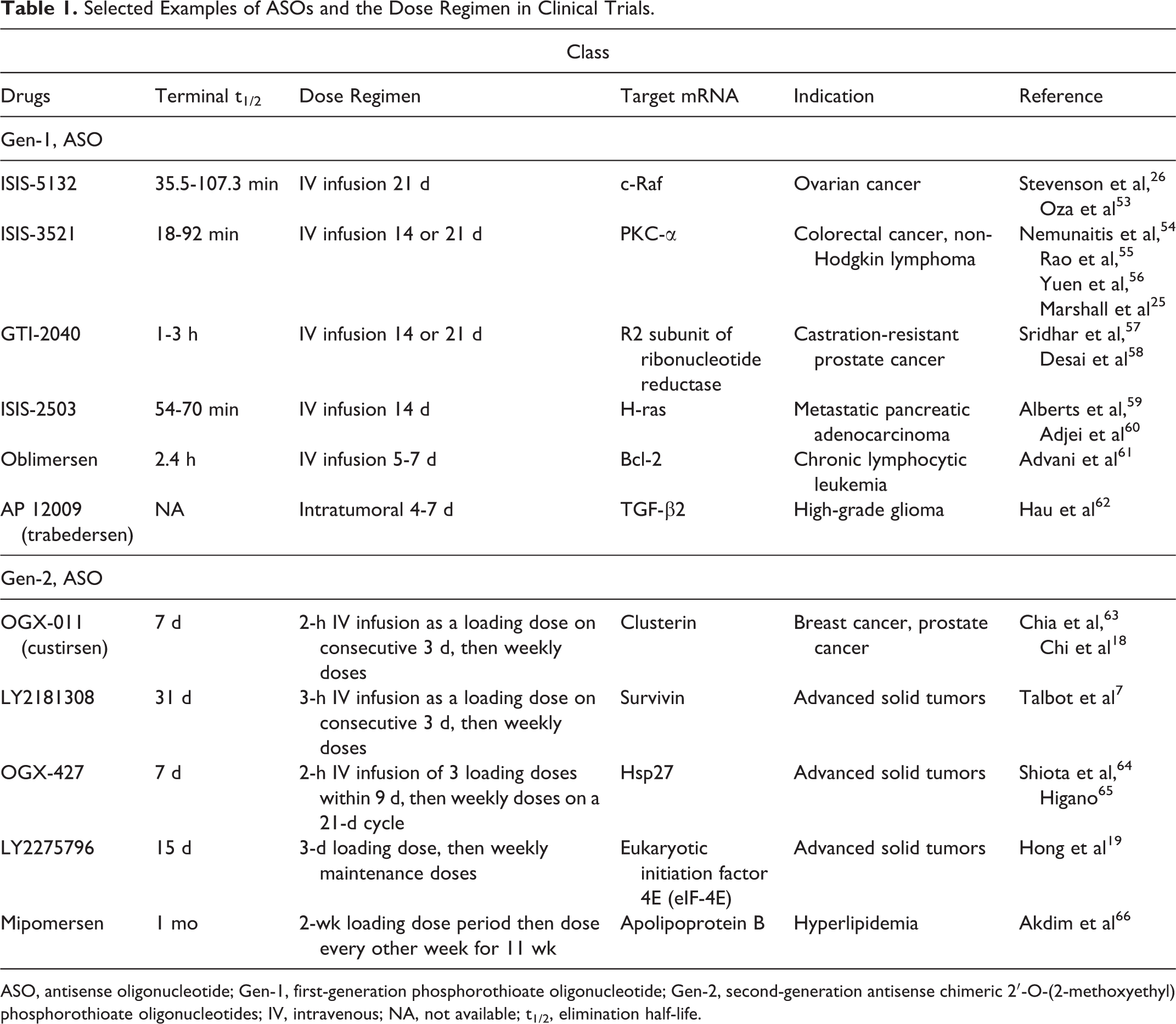
ASO, antisense oligonucleotide; Gen-1, first-generation phosphorothioate oligonucleotide; Gen-2, second-generation antisense chimeric 2′-O-(2-methoxyethyl) phosphorothioate oligonucleotides; IV, intravenous; NA, not available; t1/2, elimination half-life.
Effect of Renal Impairment on PK
The effects of renal impairment on OGN exposure have been reported. For example, an increase in the exposure of pegaptanib, a pegylated anti-VEGF aptamer, was observed with increasing renal impairment. 6 As these studies showed, the mean pegaptanib area under the curve (AUC) for patients with severe renal impairment is 37.8 μg/h/mL, which represents a 41.5% and a 60.2% increase, respectively, as compared with the patients with moderate and mild renal impairment. Similarly, the mean pegaptanib: maximum concentration (Cmax) value for patients with severe renal impairment is 96.8 ng/mL. This value represents 18.6% and 41.5% increases, respectively, as compared with patients with moderate and mild renal impairment.
A second example of the effects of renal impairment relates to LY2181308, a second-generation ASO to modulate the expression of survivin. The data from a phase 1 study in advanced cancer patients demonstrated that the LY2181308 predose or trough concentrations, as measured on days 15 and 22 in tumor tissues, were increased in patients with lower creatinine clearance. 7 While this observation is based on data from only 4 patients with the lowest creatinine clearance (<60 mL/min) in that study, it suggests a trend for higher drug exposure in tissues in these patients, as measured by the trough levels.
In a population PK analysis, creatinine clearance was found to affect mipomersen clearance but not to a clinically significant extent. Analysis of steady state dose-normalized trough concentration (Ctrough) data around week 13 showed that subjects with mild and moderate renal impairment have approximately 36% and 14% increases in mean trough concentrations, respectively. 8 While differences in PK or efficacy may not be clinically meaningful, no data on severe renal insufficiency are available. Since mipomersen undergoes urinary excretion and increases the risk for proteinuria, the European Medicines Agency recommends that mipomersen be contraindicated in severe renal insufficiency and used with caution in patients with mild to moderate renal insufficiency. The FDA-approved labeling states that mipomersen is not recommended for patients who have severe renal impairment or clinically significant proteinuria or who are on renal dialysis, owing to the lack of clinical data and renal safety profiles.
Effects of Drug-Drug Interactions on PK
OGNs are degraded by nucleases, which are ubiquitously expressed in tissues. As such, cytochrome P450 (CYP) enzymes are unlikely to be involved in OGN metabolism; CYP-mediated effects of other therapeutic agents on OGN PK are therefore predicted to be minimal. 9 The available clinical information suggests limited PK drug-drug interactions (DDIs) between OGNs and small molecular drugs. 10 In a DDI study for mipomersen, coadministration of mipomersen with simvastatin and ezetimibe resulted in a reduction in the Cmax and an increase in the area under the AUC for simvastatin and ezetimibe. 11 These changes were not considered to be clinically relevant, and thus no dose adjustment is needed. A study by Li et al 12 reported a lack of PK and PD interactions between mipomersen and warfarin. Current information on mipomersen also indicates that it causes no inhibition or induction in CYP enzymes.
A few studies have reported potential DDIs for OGNs without a clear mechanistic basis. In a trial on cancer, oblimersen, a Bcl-2 ASO, was found to increase the exposure of mitoxantrone in patients with metastatic hormone-refractory prostate cancer and decrease the formation of SN-38, an active metabolite of irinotecan. 13 Another DDI example concerns GTI-2040, a 20-mer OGN targeting the small subunit component (R2) of ribonucleotide reductase. In a study by Shibata et al, 14 the exposure of GTI-2040 was higher when the dose of the combination drug was reduced.
Some OGNs containing unmethylated CpG motifs within the sequence can stimulate immune responses, affecting the expression of host-defense proteins such as interferons and interferon-inducible genes. 15,16 Cytokines, such as interleukin 6, may affect the oxidative metabolic process by modulating the hepatic microsomal CYP isozymes and thereby potentially changing the metabolism of concurrently administrated drugs. Although OGNs are highly bound to plasma proteins, DDIs based on protein binding are expected to be minimal. 5,17
Pharmacokinetics/Pharmacodynamics
To accelerate drug development, integrated PK/PD modeling and simulation provide a promising tool to characterize the dose-exposure-response relationships and the identification of significant covariates that explain the inter- and intraindividual variability in exposure and response. One potential use of PK/PD modeling for the development of OGNs is the quantitative assessment of their PK characteristics. Unlike most small molecule drugs where renal and hepatic metabolic pathways are the major routes of elimination, OGNs are mainly eliminated by intracellular nuclease cleavage and catabolism. Since intracellular metabolism of an OGN could represent a substantial fraction to the total body clearance, PK models can describe the distribution of the drug in the extra- and intracellular spaces and systemic excretion.
Potential PD endpoints, including downregulation of mRNA or reduction of protein expression, can be useful in characterizing the PK/PD relationship and guiding the dose selection of OGNs in clinical trials. 18 –20 Various studies have demonstrated the antisense effect of ASOs and the effective reduction of target expression. 13,19,21 Pharmacologic effects of ASOs are certainly related directly to drug concentrations in the target organ. Studies have shown that ASO plasma concentrations in the postdistribution phase correlate with target tissue concentrations and can serve as a surrogate measure of target tissue concentrations for further correlation analysis with PD endpoint. 22,23
In addition to analysis of plasma and tissue levels, suborgan fractionation PK/PD studies demonstrated a dose-dependent mRNA reduction and linear PK/PD model adaptation for a second-generation ASO. 24 While these data provide insights into the relationship between drug levels and action, challenges remain in the application of the PK/PD modeling and simulation approaches. These issues relate to the achievement of effective intracellular concentrations and the relationship of intracellular concentrations to the drug plasma levels, PD response, clinical efficacy, and safety. 21,25,26
Immunogenicity
Evaluation of immunogenicity potential is generally required for a biologic product since the induction of antibodies to the product (or a related molecule) or another immunologic response may affect the action of the product and/or induce undesirable side effects. 27 While immunogenicity has been extensively investigated for proteins and peptides, OGNs represent novel structures whose potential immunogenicity has thus far received only limited study in animals or treated patients.
OGN are, by design, structurally related to nucleic acids, especially DNA, which can display surprising immunologic activity when it leaves its usual nuclear location and circulates in the blood. 28 Thus, depending on specificity of any antidrug antibody and the actual structural resemblance of an OGN to DNA, an antibody to an OGN could cross-react with endogenous DNA. 29 –32 DNA is present in the blood of normal individuals, although levels can rise in disease states characterized by inflammation or cell injury and death. 33,34 While OGNs display certain molecular features in common with DNA, these compounds have backbone modifications to modulate half-life or promote intracellular action. Importantly, these modifications could influence immunologic properties of the product, leading to differences from those of conventional DNA. These modifications could either increase or decrease immunologic activity. As demonstrated in studies with synthetic and natural nucleic acids, the immune properties of DNA are determined by base sequence, base modification (ie, methylation), strandedness, and backbone modification. As a result, DNA can exist in stimulatory, inhibitory, or inert forms. 35,36 Protein binding can also influence the immunologic profile of DNA by changing the cellular uptake and trafficking. The proteins that can modify the immune activity include anti-DNA antibodies and DNA-binding proteins such as LL-37, a neutrophil product known as a cathelicidin. 37,38
Depending on its physical-chemical properties and protein association, DNA can stimulate immune responses by triggering internal DNA sensors, most prominently toll-like receptor 9. These internal sensors have evolved most likely to respond to bacterial or viral DNA present in the cytoplasm of cells during infection. 39 –41 In view of the signaling properties of these sensors, modifications of DNA that affect cellular uptake and intracellular persistence could affect receptor stimulation. In the case of an immunomodulatory OGN, for example, this stimulation is the goal of the product, although inhibitory OGNs have also been explored for use in the setting of inflammatory and autoimmune disease. 35,36 A further consideration about immunogenicity of OGN concerns the relationship of any induced antibodies to autoimmunity. Antibodies to DNA are the serologic hallmark of systemic lupus erythematosus, a prototypic systemic autoimmune disease. Anti-DNA antibodies can bind to both double- and single-stranded DNA, primarily recognizing conserved structures on the phosphodiester backbone. 42,43 By forming immune complexes with endogenous DNA, these antibodies can promote pathogenesis by depositing in the kidney to stimulate inflammation; these complexes can also stimulate cytokine production, in particular type 1 interferon by plasmacytoid dendritic cells. 44,45
As OGNs have some structural similarity to DNA, induced antibodies could induce responses if they cross-react with endogenous DNA. Studies suggest that anti-DNA antibodies from lupus patients can bind to phosphorothioate OGNs even though preferred antigens for DNA with a phosphodiester backbone are usually much larger; a large piece of DNA is needed since anti-DNA antibodies bind to DNA by a mechanism called monogamous bivalent interaction, which requires pieces of DNA 40 to 50 nucleotides or longer. 46,47 Because the phosphorothioate backbone has significantly enhanced protein binding, 48 an OGN—even if small (eg, 20-30 nucleotides)—could bind to an anti-DNA.
In assessing potential cross-reactivity to DNA of antibodies induced to an OGN, the choice of the anti-DNA assay is important. Current anti-DNA assays vary in antibody measurements with patient sera, reflecting a range of anti-DNA antibody specificities detected under the assay conditions. For example, assays commonly used in the clinical arena differ in detection of high- vs low-avidity antibodies, and a sera positive in one assay may be negative in another. 49
At this time, the assay formats most appropriate for screening any potential cross-reactive anti-DNA binding of an anti-OGN are not known; another issue concerns assay conditions. The binding of lupus anti-DNA antibodies can be very salt dependent because of the importance of electrostatic interactions in antibody binding. 50 To the extent that the binding of antibodies to an OGN is also electrostatic, high-salt conditions may limit antibody assessment with OGN antigens.
Several mechanisms may thus affect the immunogenicity of OGN-based therapeutics: (1) the use of modified analogs designed to increase nuclease resistance, (2) linking an OGN to a protein carrier to limit nuclease digestion and enhance cellular uptake, and (3) encapsulation in a pegylated liposome.
Although polyethylene glycol (PEG) is generally regarded as nonimmunogenic, administration of pegylated products can lead to antibody responses. This situation is evident in studies of the induction of IgM and IgG antibodies against the PEG component of the vehicle by the activation of B cells that take up the stimulatory siRNA. 51 The immune response against delivery vehicles containing siRNA generated sufficient titers of serum antibodies in mice to increase blood clearance, decrease efficacy, and induce hypersensitivity reactions on drug readministration.
Immunogenicity Case Study: Mipomersen
Mipomersen sodium, an ASO that inhibits the synthesis of apolipoprotein B-100, was approved by the FDA for treatment of homozygous familial hypercholesterolemia. Mipomersen is a 20-base synthetic OGN sodium salt and a first-in-class antisense inhibitor of apolipoprotein B-100 synthesis. Approximately 37.5% of the phase 3 patients were positive for anti-mipomersen antibodies, and 72% of patients were anti-drug antibody positive in the extension trials. 52 The investigator concluded that antidrug antibodies can interfere with the bioanalytical assay methodology; therefore, mipomersen exposure data from these clinical trials may not be reliable. 52 The incidence of neutralizing antibodies has not been reported, and the effects of antibodies on PK, efficacy, and safety are unknown.
Summary
While the development of ASO therapies has been impressive, structural resemblance of these compounds to nucleic acids and their intracellular mode of action raise important challenges in clinical pharmacology. Given the immunologic properties of nucleic acids, the significance of immunogenicity may extend beyond effects of the production of antibodies that affect drug efficacy/safety or alter PK. The development and possible consequences of immunogenicity may need further investigation in the future clinical development of OGN-based therapeutics.
Footnotes
Acknowledgments
We thank Drs Shiew-Mei Huang and John Leighton at the FDA for their valuable comments and critical review of the manuscript.
Declaration of Conflicting Interests
Dr. Pisetsky has consulted with Genzyme and ISIS Pharmaceuticals on the subject of oligonucleotide immunogenicity.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was partially supported by the US FDA Regulatory Science and Review Enhancement funding and fellowship support for Dr Lon from Oak Ridge Institute for Science and Education.
Author Notes
This article reflects the views of the authors and should not be misconstrued as representing FDA’s views or policies. The content has been partially presented at the AAPS National Biotechnology Conference, San Francisco, CA, 2011; DIA/FDA Oligonucleotide-Based Therapeutics Conferences, Washington, DC, 2012; and DIA/FDA Oligonucleotide-Based Therapeutics Conferences, Washington, DC, 2013.