
Abstract
Background:
A significant number of inquiries are issued during the review of pharmaceutical products developed using the quality-by-design (QbD) approach in Japan. The purpose of this article was to identify key elements specific to QbD development that should be described in the Quality Overall Summary (QOS) dossier.
Methods:
The review reports of the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) were investigated to identify review points for QbD products. Based on the results, key considerations for preparing the QOS of QbD products were determined.
Results:
The primary review points for QbD products were control strategy (∼30%), design space (∼15%), and real-time release (∼15%). Additionally, the market authorization application form (AF) was discussed more frequently for QbD products than for products developed using a traditional approach. Based on the results, a “QOS checklist for QbD products” was developed to highlight key elements of QbD products that should be included in the QOS.
Conclusions:
It is important to explain the scientific rationale for the control strategy, design space, and real-time release in the QOS and how “regulatory flexibility” is expressed in the AF. The QOS checklist enables applicants to prepare an appropriately detailed QOS that should satisfy the PMDA’s critical review points. The following are recommended for further discussion topics to enable efficient and consistent review by the PMDA for QbD products: (1) Clear guidance on how to express “regulatory flexibility” in the AF and (2) a “QOS checklist for QbD products” that is agreed upon by both regulatory agencies and pharmaceutical companies.
Keywords
Introduction
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) has recently developed harmonized guidelines for drug development. Based on the ICH guidelines, pharmaceutical product development data may be incorporated in global marketing applications. Consequently, pharmaceutical products are increasingly developed with a goal to achieve global simultaneous submissions. 1 In addition, pharmaceutical companies are encouraged to use an enhanced quality-by-design (QbD) approach to facilitate the implementation of robust manufacturing processes that reliably produce high-quality pharmaceutical products. 2
QbD is defined as “a systematic approach to development that begins with predefined objectives and emphasizes product/process understanding and process control based on sound science and quality risk management.” 3 Under the QbD paradigm, product quality is assured by not only end product testing but also process controls that are defined based on process understanding and risk management. 4 The QbD development approach enables better process understanding and the establishment of a robust manufacturing process. In addition, QbD principles enable the use of design spaces that allow pharmaceutical companies to apply a more flexible regulatory approach toward changes in the manufacturing process. A design space is a variable space comprising input variables and process parameters that has been demonstrated to provide assurance of product quality 3 and can be established based on the results of design of experiments (DoE) and statistical analyses. Furthermore, a real-time release (RTR) strategy based on thorough process controls via process design and/or process analytical technology may reduce the costs of release tests and/or supply lead time, resulting in inventory reduction. Thus, the QbD approach can significantly benefit pharmaceutical companies from the perspectives of quality, cost, and regulation. 5
From 2008 to 2014, a total of 268 pharmaceutical drugs containing new chemical/biological entities or vaccines were approved in Japan. Among the approved drugs, 33 were developed using a QbD approach for drug substance and/or drug product manufacturing processes. The percentage of products applying a QbD approach relative to the approved new drugs has been increasing over the past 2 years in Japan (Figure 1). Note that products developed using a QbD approach (QbD products) are defined as products applying a QbD approach as evidenced in the review reports or other public information of the Japanese Pharmaceuticals and Medical Devices Agency (PMDA).
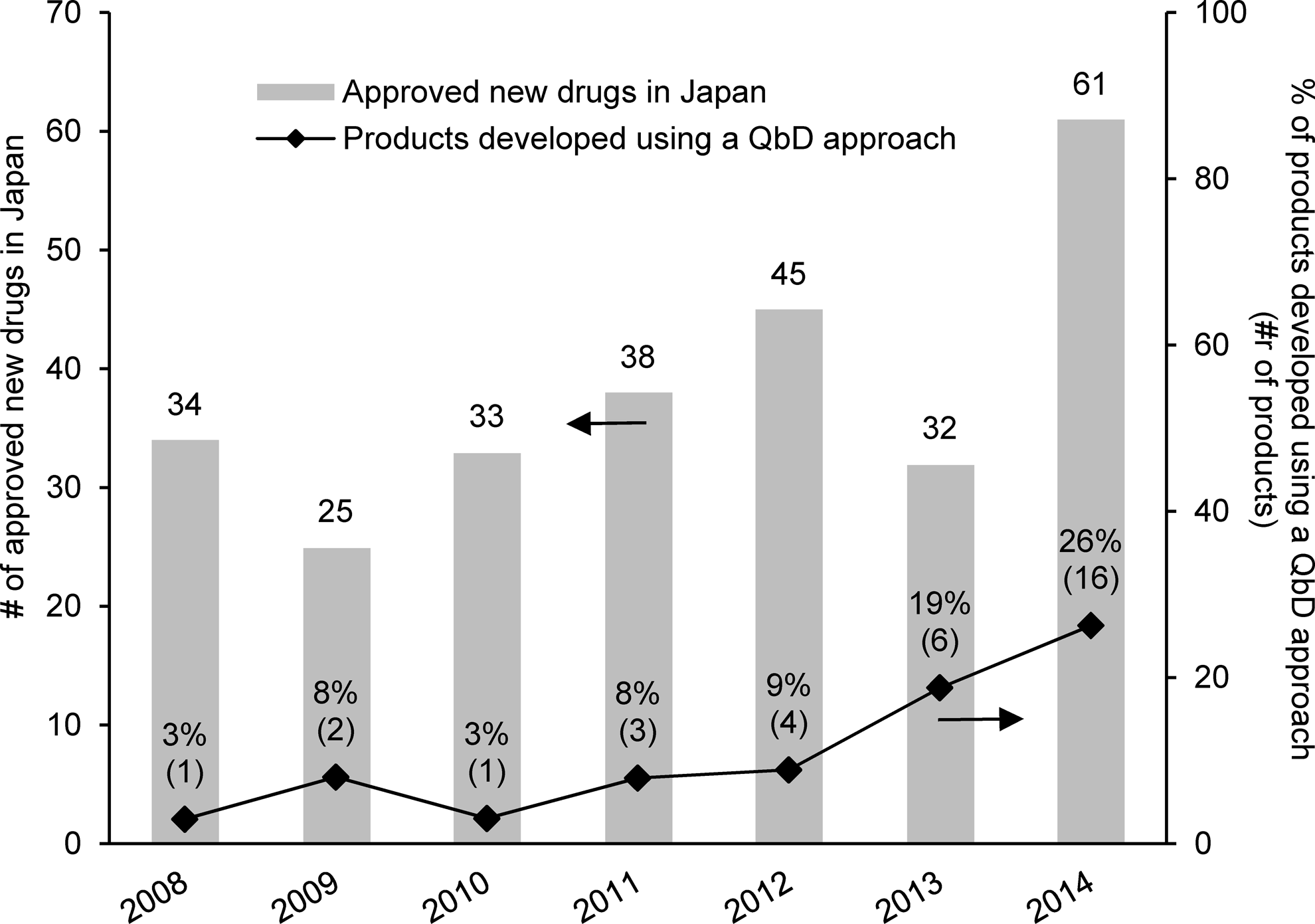
Percentage of products using a quality-by-design approach among approved pharmaceutical drugs containing new chemical/biological entities or vaccines in Japan (2008 to 2014).
The ICH has issued “Q8/Q9/Q10 Points to Consider,” 6 and a Japanese Ministry of Health, Labor and Welfare (MHLW)–sponsored science research study group has also published case studies to present clear interpretations of QbD-related guidelines. 7 –9 However, the PMDA’s review expectations for QbD products remain unclear to pharmaceutical companies, and a significant number of the PMDA’s inquiries are issued during the review of QbD products. It is important for applicants to understand how a significant amount of development data generated using a QbD approach should be summarized in the dossier. Because the Quality Overall Summary (QOS) is the primary review document in Japan, the content of the QOS is a critical factor for rapid and efficient review by the PMDA. 10 Thus, the purpose of this article is to define key elements to describe the QOS for QbD products.
Materials and Methods
After 2012, if the PMDA concludes that a manufacturing process is developed using a QbD approach, this information is disclosed in the PMDA’s review reports, particularly in the <Summary of the submitted data> section. Products for which a QbD approach is not directly stated in the report or other public information are assumed to be developed using a traditional approach (non-QbD products). The PMDA’s critical inquiries, the sponsor’s responses to the PMDA inquiries and the PMDA’s final position are located in the <Outline of the PMDA review> section. In this study, the PMDA’s review reports for new drugs approved from 2008 to 2014 in Japan were reviewed. The PMDA’s comments/questions (which are listed in <Outline of the PMDA review>) were classified into several categories (control strategy [eg, process controls and process understanding], design space, RTR, specification and test methods, stability, novel excipients, others, and no specific comments) based on the context of the discussion. In addition, specific PMDA review points for QbD products were identified. Key elements to consider for the QOS preparation of QbD products are discussed based on these results.
Results
A thorough investigation of the <Outline of the PMDA review> section indicated that the PMDA’s review points for QbD products were specifically related to “control strategy” (29.4% of the PMDA’s total comments/questions for QbD products), “design space” (14.7% of the PMDA’s total comments/questions for QbD products), and “real-time release” (14.7% of the PMDA’s total comments/questions for QbD products), whereas the review points were related to “specification and test methods” (28.6% of the PMDA’s total comments/questions for non-QbD products) and “stability” (23.1% of the PMDA’s total comments/questions for non-QbD products) for the products developed using a traditional approach (Table 1).
The PMDA’s Review Points for Products Developed Using a Quality-by-Design (QbD) Approach and a Traditional Approach.
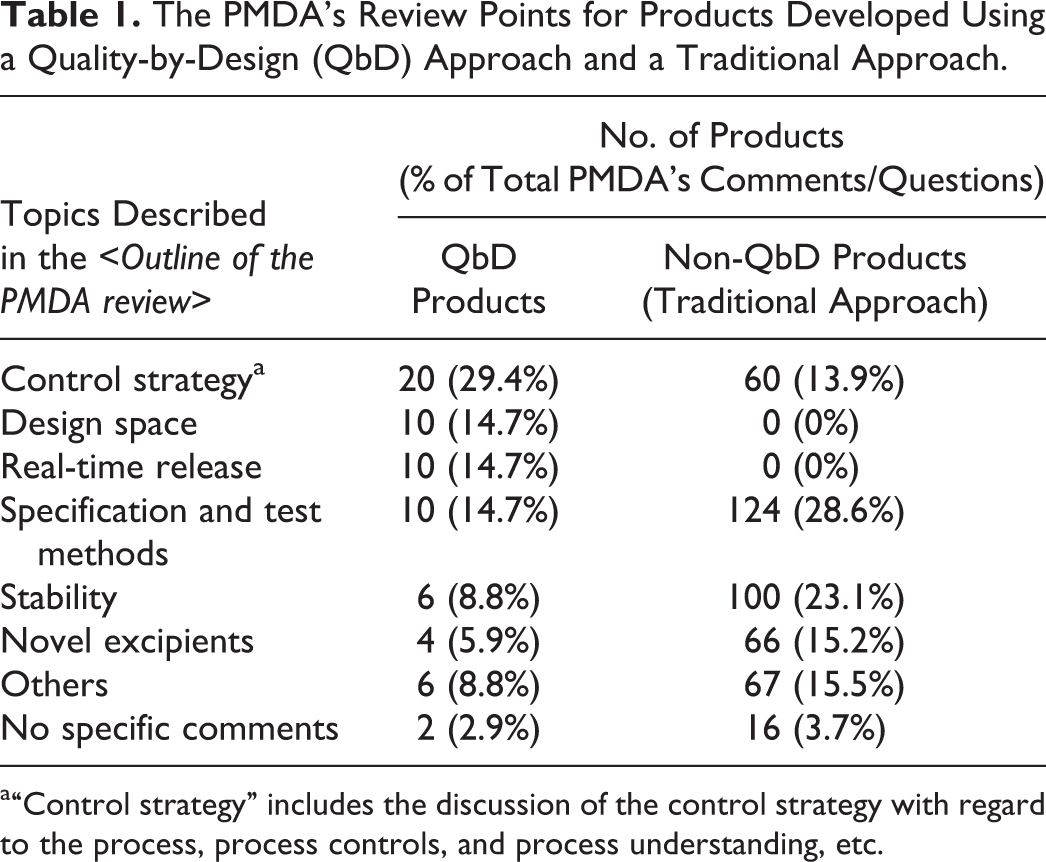
a“Control strategy” includes the discussion of the control strategy with regard to the process, process controls, and process understanding, etc.
The PMDA’s Review of QbD Products
Control strategy
The most common category of review comments described in the review reports for QbD products were related to “control strategy.” The PMDA’s major review comments for control strategy are summarized in Table 2. There were inquiries into the justification of critical quality attributes, critical process parameters, and critical processes, and the PMDA has also asked how the identified critical process parameters were controlled in the process. The main review points included the development history of the control strategy and its justification. In addition, specific to QbD products, there was significant discussion between the PMDA and applicants for the description of a market authorization application form (AF). Note that the AF is the legally binding portion of the dossier in Japan and is subject to regulatory approvals if any attributes are changed. There are 2 categories of postapproval changes in Japan. 11 For minor changes akin to the United States Food and Drug Administration (FDA) “changes being effected,” a minor change notification (MCN) is required, and an applicant must submit the amended AF to the PMDA of the change within 30 days of implementation. For major changes akin to the FDA “prior approval supplement,” a partial change application (PCA) is required that includes an updated AF and/or QOS. The PCA has a high regulatory burden and generally requires 6 to 12 months for approval. The following 2 items were major discussion points related to the AF: (1) how much information on the process description (eg, process parameters) should be provided in the AF and (2) the justification for MCN or PCA items (which should be based on the results of risk management). Furthermore, the PMDA indicated that for some products, the terminology used in the dossier did not follow the ICH definition, particularly when discussing critical quality attributes. The PMDA clearly stated in the review report that “quality attribute criticality is primarily based upon severity of potential impact on the efficacy/safety and does not change as a result of control factors.”
The PMDA’s Review Comments for Control Strategy (Major).

Design space
Among the 33 QbD products, 9 approved products developed design spaces for their manufacturing processes. The PMDA’s major review comments for design spaces are summarized in Table 3. The discussion points for the design spaces primarily involved the development history of the design spaces and their justification. For one product, the DoE and the development of the design space were elaborated in the review report. This elaboration implies that there was a significant amount of communication on the design space between the PMDA and the applicant during the review. In addition, similarly to the review comments on the control strategy, there were review comments on the AF stating that (1) the AF must clarify which parameters are components of the design space and identify the mutual interactions of these parameters in that design space and (2) a design space that ensures a critical quality attribute should not be classified as an MCN item, even if the parameters are strictly controlled around the target. This investigation revealed that there are specific expectations in the AF on the design space and whether the established design space is classified as an MCN or PCA item.
The PMDA’s Review Comments for Design Space (Major).

Real-time release
Among the 33 QbD products, nine approved products have employed RTR for a drug substance or drug product release. The PMDA’s major review comments for RTR are summarized in Table 4. When an in-process test or intermediate test was determined to be used for RTR, justifications of the substitution of alternate test results for the final product release testing results were requested. In addition, for the limited number of products that established a mathematical model for RTR, a detailed explanation was requested, including how to develop the prediction model and the justification of the model (eg, model performance for commercial-scale products, discrimination power for defective products).
The PMDA’s Review Comments for Real-Time Release (RTR) (Major).

Discussion
Key Elements of the QOS for QbD Products
A large amount of experimental data and statistical analyses are obtained during QbD development to understand the process. It is thus important to provide a holistic summary that demonstrates a complete understanding of QbD development in the QOS (CTD Module 2.3) to support an efficient review as a part of a CTD review or GMP compliance review. However, this study determined that no satisfactory information on control strategy, design space, or RTR was provided in the QOS. Alternatively, it may be difficult to understand the sponsor’s QbD approach from the QOS based on the extent of inquiries from the PMDA in their review reports.
Notably, during QOS preparation, applicants must acknowledge the definitions of QbD-related terminology that are specified in ICH Guideline Q8 (R2). If the definitions of terminologies differ between reviewers and applicants, there may be a significant discrepancy during the discussion of product quality. Specifically, applicants should pay attention to the definitions “Critical Quality Attribute: Quality Attribute criticality is primary based upon severity of harms and does not change as a result of risk management” and “Critical Process Parameter: Process Parameter criticality is linked to the parameter’s effect on any critical quality attributes. It is based on the probability of the occurrence and on any detectability, and therefore can be changed as a result of risk management.” 3,6 If applicants utilize terminology that is not defined in the ICH guidelines, the relevant definition should be clearly specified in the QOS.
Control strategy
For QbD products, the justification of the established control strategy was the main discussion topic during PMDA review. Because the development approaches for a drug substance or drug product vary depending on the company and individual compound, an overall picture of the QbD development strategy is recommended as an introduction to enable the reviewers to easily understand the development history of the control strategy. In SAKURAKAIKA Tablets mock P.2, which was issued by the MHLW-sponsored science research group, 9 an overall picture of QbD development was presented at the start of section P.2.3 (drug product manufacturing process development). In addition, specific lists of critical quality attributes and critical process parameters are required in the QOS along with the justifications, which are based on prior knowledge/experience, DoE results, and risk assessment. Furthermore, the control strategy, including the control of raw materials, the control of critical process parameters, in-process testing, and design space, that ensures the identified critical quality attributes, should be clearly presented based on the results of the experiments and risk management strategies.
Design space
When a design space is established to ensure critical quality attributes, the detailed results of the DoE that were executed to construct the design space were discussed in the review report. The DoE used to develop the design space, the actual experimental results of the DoE, and the statistical analyses (which form a basis for the design space) should be presented in the QOS. In addition, the PMDA questioned whether or not the design space was applicable at a commercial scale. When the design space is developed using data from a laboratory scale or pilot plant scale, the potential risk of the scalability of the design space should be considered and discussed in the QOS.
Real-time release
The use of RTR must be justified. When the prediction values from the mathematical model are used for the release of products instead of end product testing, the scientific rationale should be explained in detail. Specifically, the lot information used for model construction (eg, whether or not lots that exhibit the profile with a specification boundary and/or that do not conform to the specifications were used in the model construction) and the accuracy between the predicted and observed values should be included in the QOS. For one product, the PMDA requested confirmation of the consistency between the actual dissolution results and the predicted values from the model for the commercial lots directly following approval. Therefore, applicants should thoroughly prepare the model performance at commercial-scale lots and the model maintenance procedures during commercial production so that responses to PMDA inquiries can be provided as quickly as possible.
Process Description in the AF
Some examples of the process description for QbD products were presented in the “SAKURA TABLET Mock Application form” 7 and “SAKURAMIL Mock Application form,” 8 which were created by the MHLW-sponsored science research group. However, the PMDA’s review reports revealed a significant number of inquiries related to the descriptions within the AF.
In “SAKURAMIL Mock,” the MHLW-sponsored science research group proposed the following justification for regulatory flexibility for QbD products: “A description of target/set values is not necessary for parameters in cases in which the enhanced approach demonstrates that there is no impact on quality.” However, the PMDA claimed in the review report that “even if the process parameters do not impact critical quality attributes in the studied range, at a minimum the AF must describe the manufacturing operation and/or process parameters that should be controlled within specified ranges to ensure the product meets the specifications” and “the information level of process parameters in the AF can be determined by the control strategy established based on risk management results. However, at a minimum, factors that are required to understand the manufacturing operation should be specified in the AF and are subject to regulatory change control.” In addition, the PMDA also stated that “regarding parameters composing the design space, it is not appropriate to specify in the AF only parameters with a relatively high impact on quality.” How much regulatory flexibility can be obtained may be dependent on the level of product understanding and scientific knowledge gained in the individual development activities; however, there is a gap between the regulatory agency and the applicants regarding how the regulatory flexibility could be realized in the AF. No clear guidance regarding how the regulatory flexibility is proposed in the AF is a regulatory problem that results in significant discussion during the review of QbD products in Japan. At present, it is important that the scientific rationale for the proposed regulatory flexibility is clearly explained in the QOS, referring to the PMDA’s positions described in the latest review reports.
Acceleration of the PMDA’s Review for Products Developed Using a QbD Approach
A significant number of inquiries are issued during the review of QbD products compared with products developed using a traditional approach to confirm the adequacy of the established control strategy and whether the control strategy is appropriately reflected in the AF. However, the total review period has been shortened over the years. 12 Consequently, it is particularly important for applicants to prepare a detailed QOS that addresses the PMDA’s review points identified herein to minimize the number of inquiries. Based on the identified the PMDA’s review points for QbD products and the relevant ICH guidelines (including a Q&A), a list of critical review points for QbD products has been summarized as a checklist entitled “QOS checklist for QbD products.” This approach is similar to the “Question-based Review for Chemistry, Manufacturing and Controls Evaluations of Abbreviated New Drug Applications” by the FDA. 13 The applicants may consider attaching responses to the categories listed in Tables 5 to 7 to the QOS so that the PMDA reviewers can easily understand the critical elements of the QbD approach. This approach also attempts to create a QOS that will satisfy the PMDA’s viewpoints. In addition, if the “QOS checklist for QbD products” can be aligned across multiple regulatory agencies, a consistent review can be achieved for QbD products. Thus, if the “QOS checklist for QbD products” agreed upon by both regulatory agencies and applicants is published, an efficient review for QbD products will be facilitated.
Quality Overall Summary Checklist for QbD Products—Control Strategy.

aThe definition of critical process should be referred to as PFSB/ELD Notification No. 0210001 dated February 10, 2005. 11
Quality Overall Summary Checklist for QbD Products—Design Space.

Quality Overall Summary Checklist for QbD Products—Real-Time Release.

Conclusion
The main review points for QbD products were related to the control strategy, design space, and RTR. This study indicates that the expected level of information related to the control strategy, design space, and RTR is not currently provided in the QOS or the PMDA has not completely understood these items from the submitted QOS. In addition, there were far more inquiries/discussion related to the AF for QbD products than for products developed using a traditional approach. Consequently, it is important to explain the scientific rationale for QbD elements, including the control strategy, design space, and RTR, in the QOS and how the “regulatory flexibility” is expressed in the AF. This study established a “QOS checklist for QbD products” to capture key elements of the QOS for QbD products. This checklist enables an applicant to create a sufficiently detailed QOS in an attempt to adequately satisfy the PMDA’s critical review points. Finally, the following topics are recommended for discussion to enable efficient and consistent review by the PMDA of QbD products: (1) clear guidance on how to express “regulatory flexibility” in the AF and (2) a “QOS checklist for QbD products” that is agreed upon by both regulatory agencies and pharmaceutical companies.
Footnotes
Acknowledgments
The authors are indebted to Jennifer A. Jakubowski, PhD, for her careful reading of this manuscript and thoughtful comments.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.