
Abstract
Background:
The use of smart and/or wearable devices for collection of electronic data in clinical trials has recently become a strong tool with which to collect patients’ data in a timely manner. Electronic collection of patient data will necessitate comprehensive data analysis involving huge-scale datasets in the future. However, it is still unclear how to validate and qualify computerized systems used to collect and/or manage electronic clinical data when smart and/or wearable devices are involved.
Methods:
We (a special interest group of Good Automated Manufacturing Practice Japan Forum [GAMP Japan]) investigated and designed a data-flow model for a clinical data management system involving smart and/or wearable devices, and suggested an approach for the validation of such a computerized system. The appropriateness of applying GAMP5 to the validation of a clinical data management system involving smart and/or wearable devices was also reviewed.
Results:
A regulated company should have policies and standard procedures for validating computerized systems in clinical systems. When a sponsor engages a contract research organization (CRO) for clinical data management, the sponsor should assess the CRO to confirm their capabilities. The sponsor also needs to check whether the CRO assesses device manufacturers as sub-suppliers. When the CRO intends to conduct sub-supplier assessment with a device manufacturer, a risk-based approach can be taken.
Conclusions:
We believe our method of system validation will be applicable to and will facilitate various clinical trials that involve smart and/or wearable devices.
Introduction
Recently it is most common for clinical trials to be conducted by using electronic trial data handling systems and/or remote electronic trial data systems. The ICH E6 Good Clinical Practice guideline stipulates that the sponsor is required to validate such electronic trial data-management systems by confirming that the system conforms to the sponsor’s requirements and is capable of performing as intended. 1 The scope and application of Part 11 of Title 21 of the Code of Federal Regulations (21 CFR part 11) stipulates that records must be maintained or submitted in accordance with the predicate rules. 2
Detailed validation procedures are not indicated in the Good Clinical Practice guidelines or in 21 CFR Part 11. In accordance with 21 CFR Part 11, procedures and controls need to be employed and designed to ensure the authenticity, integrity, and the confidentiality of electronic records. 3 Procedures shall include system validation, generation of copies of records, record retention, system access controls, time-stamped audit trails, and education/training of personnel. 3
When it comes to clinical trials, computerized systems should be designed “to prevent errors in data creation, modification, maintenance, archiving, retrieval, or transmission” as written in the US Food and Drug Administration (FDA) Guidance for Industry: Computerized Systems Used in Clinical Investigations. 4 Users of clinical systems should establish and maintain standard operating procedures, and retain source documents and/or data. The system should have functions to control security through access limitation and the ability to create audit trails.
The International Society for Pharmaceutical Engineering (ISPE) is a worldwide association that deals with regulatory, scientific, and technical topics focusing on pharmaceutical engineering. ISPE members play active roles in each region. The Good Automated Manufacturing Practice Japan Forum (GAMP Japan) is one of the Japanese Communities of Practice of the ISPE and comprises several special interest groups. Because topics related to clinical systems also fall within the scope of the GAMP Japan Forum, a special interest group was formed in 2009 to discuss validation of clinical systems. At that time, the group comprised 9 members, including clinical specialists from pharmaceutical industry, consultants, and information technology (IT) suppliers.
The group classified clinical systems into 4 types based on their intended purpose: (1) clinical data management systems (CDMSs), (2) electronic data capture systems offered by application service providers (ASP-modeled EDCs), (3) statistical analysis systems, and (4) clinical trial management systems (CTMSs). 5
Some discussion has been published on validation methodologies for traditional computerized systems. 6,7 In the scope and application of 21 CFR Part 11, it is recommended to refer to industry guidance such as the GAMP4 Guide for practical guidance on the validation of computerized systems. 2 In 2008, 5 years after the scope and application of 21 CFR Part 11 was issued, GAMP5 was published. 8 GAMP5 illustrates various validation approaches based on software categories: Category 1, infrastructure software; Category 3, nonconfigurable software including commercial off-the-shelf (COTS) software; Category 4, configured software; and Category 5, custom applications. (Category 2 no longer exists in GAMP5; Category 2, firmware, was formerly included in the system classification list in GAMP4.)
CDMSs, ASP-modeled EDCs, and CTMSs are set up for each clinical trial with some configuration adjustments and can be applied to production with appropriate configuration management. Thus, those systems would be validated by taking a Category 4 approach and continuing change management. 5 Statistical analysis systems would be validated by taking a Category 5 approach because each program is fully customized. 5 That is to say, GAMP5 can be applied to computerized system validation in clinical systems.
Smart devices and wearable devices have penetrated the clinical arena and are positioned as handy tools with which to collect and evaluate various clinical observations such as pulse, blood pressure, eye pressure, and blood sugar. For instance, wearable trackers for upper-arm blood pressure enable repeated evaluation of blood pressure easily. 9 Blood pressure is one of the most important parameters used to evaluate treatment efficacy in cardiovascular diseases. Smartphones are used more often to collect patient-reported outcomes. 10 –12 For example, patients with Parkinson’s disease may input their emotional conditions into their smartphones by answering short daily questionnaires, and investigators can browse a patient’s input and easily know their day-to-day conditions.
Such devices are available to evaluate drug efficacy in some clinical trials. When clinical data are collected through devices from patients directly, such clinical systems need to be validated, because the integrity of such data impacts highly on the clinical evaluation of the medicinal product in question.
Electronic patient data collection will necessitate comprehensive data analysis involving huge-scale datasets in the future. The Precision Medicine Initiative Cohort Program, for which developmental planning has been underway since 2015 in the United States, is going to be carried out with patients’ voluntary participation to measure risks of diseases related to environmental triggers, genetic factors, and so on. 13 Leveraging electronic health records and health infrastructure is also within the scope of that program.
Although validation of computerized systems is required in the regulatory rules and by the Good Clinical Practice guidelines, it has not been determined how smart devices and wearable devices should be validated. Medical devices themselves may be qualified or authorized by health authorities, 14 –16 and not be subject to the process of computerized system validation. Validation practitioners are unsure about how to validate computerized systems involving smart devices and/or wearable devices as clinical measurement tools, but sponsors who conduct clinical trials have a responsibility to ensure the integrity of the clinical data collected through such systems.
The objective of this study was to design a clinical data flow model in a clinical data management system involving smart and/or wearable devices and to determine procedures to validate such a system. This study does not contain any studies with human or animal subjects performed by any of the authors.
Methods
A special interest group of GAMP Japan focusing on validation of clinical systems involving smart and/or wearable devices was newly formed in 2014, with 6 members, including clinical experts from the pharmaceutical industry, consultants, academics, and IT suppliers. Meetings of the special interest group were held every month from January 2014 to August 2015; there were a total of 18 regular meetings and 3 ad hoc meetings.
In the first meeting and in several subsequent meetings, a literature review was conducted. Relevant regulatory requirements, legislation, and guidance from health authorities such as the US FDA were investigated. Because clinical data management processes were not clearly documented in any published articles, a clinical data flow model was discussed and designed by considering regulatory rules and by clarifying the roles and responsibilities of key players working in clinical data management systems involving smart and/or wearable devices.
The scope of discussions at the special interest group meetings was trials sponsored by a regulated company and aimed at the registration of a new drug or indication. In general, such registration trials are well controlled and conducted with standardized procedures where the roles of the key players are defined. On the other hand, nonregistration trials are operated differently by regulated companies or physicians. We focused on registration trials and discussed typical models of registration trials with a data collection system involving smart devices and/or wearable devices.
A general clinical data flow model was discussed, instead of a model specific to a particular medicinal product.
The appropriateness of applying GAMP5 to the validation of clinical data management systems involving smart and/or wearable devices was also reviewed by considering the following points: (1) how electronic data management systems involving smart devices and/or wearable devices differ from legacy data management systems without such devices, (2) how to conduct supplier assessment, and (3) how to take a risk-based approach. Other considerations were also noted.
Results
Over the period from January 2014 to August 2015, the special interest group held 18 regular meetings and 3 ad hoc meetings. The amount of meeting time was more than 50 hours in total. Various clinical trial models involving different types of wearable devices were investigated and discussed by considering the different regulatory requirements of the United States and Japan.
Characteristics of Electronic Data Management Systems Involving Smart Devices and/or Wearable Devices
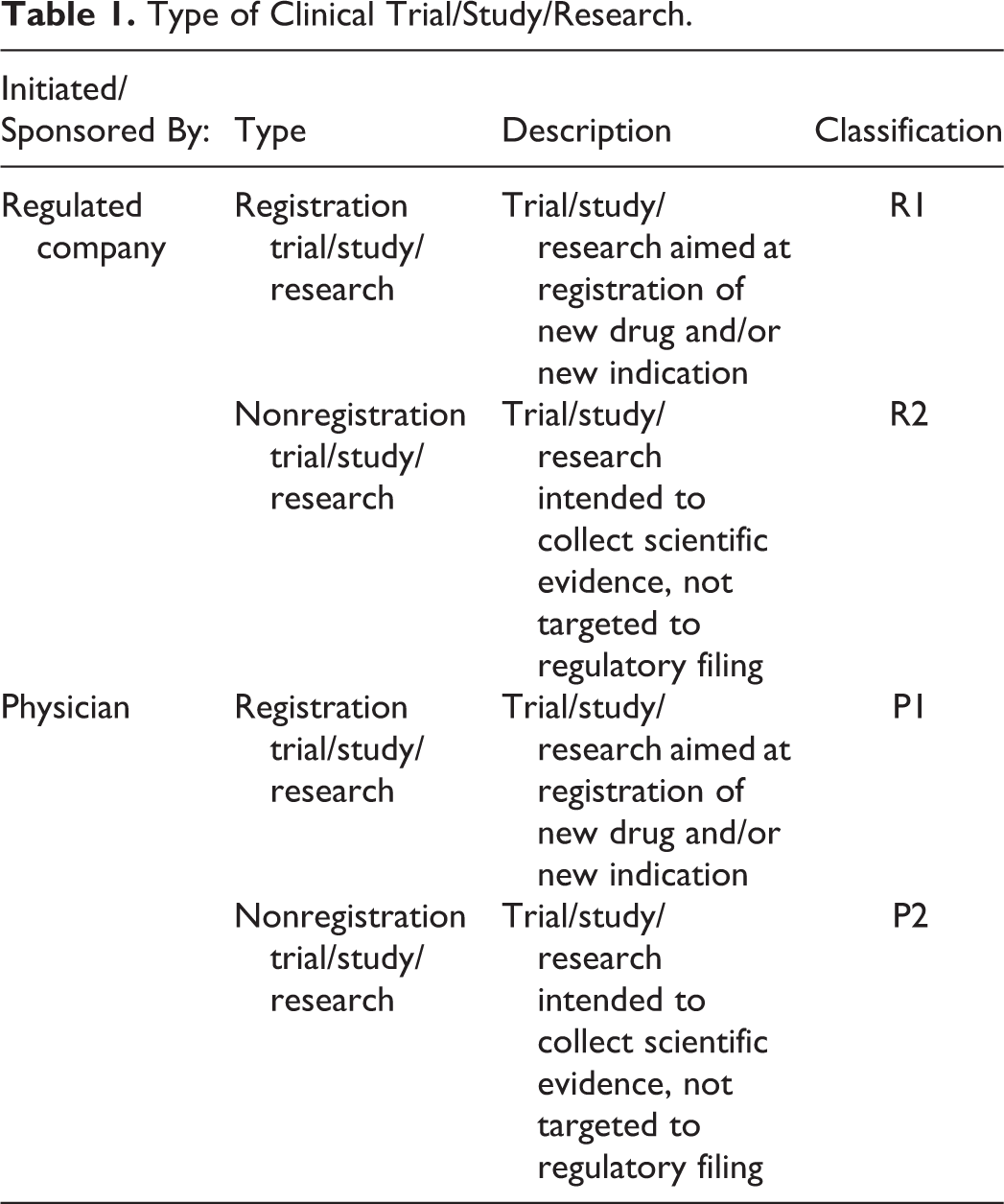
Clinical trial/study/research was classified into 4 types (R1, R2, P1, and P2) based on the type of sponsorship and intended outcomes (Table 1). The scope of the special interest group discussions was registration trials/study/research, R1.
Type of Clinical Trial/Study/Research.
Roles and responsibilities are assumed to be as shown in Table 2. We focused on the case where the sponsor engages a contract research organization (CRO) for its trial operations, and the clinical database server is available via a cloud computing service provided by the CRO. Trial conduct and data management are dealt with by a CRO under a written agreement with the sponsor. The clinical data flow model of the electronic data management system involving smart and/or wearable devices was designed as shown in Figure 1. The CRO provides the sponsor with a clinical data management system which enables data collection and data cleaning, as well as system operation support services. The CRO employs smart and/or wearable devices developed and delivered by manufacturer(s). Investigators who join the clinical trial can browse such patient-reported outcomes kept in the central server. They can also enter patient data electronically upon patients’ site visits. The sponsor may browse the data collected with devices from patients or entered by the investigators.
Roles and Responsibilities of Key Players Working in Clinical Data Management Systems Involving Wearable and/or Smart Devices.
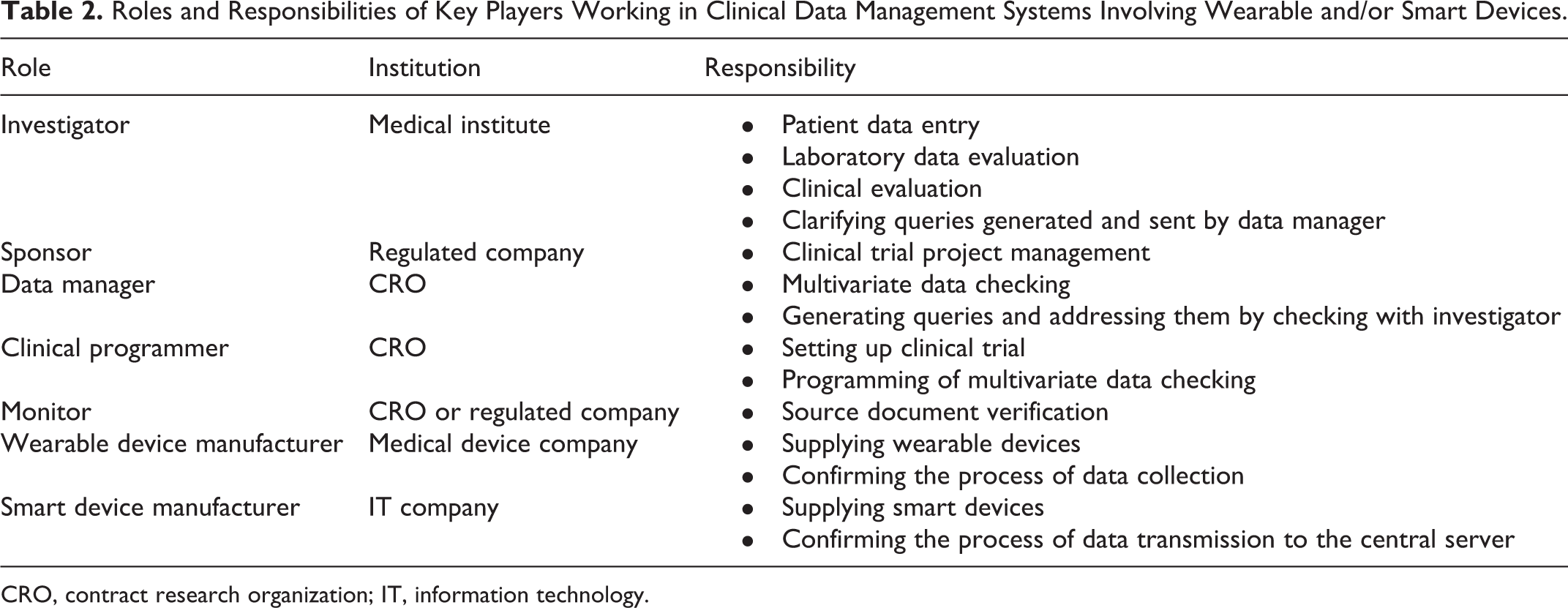
CRO, contract research organization; IT, information technology.
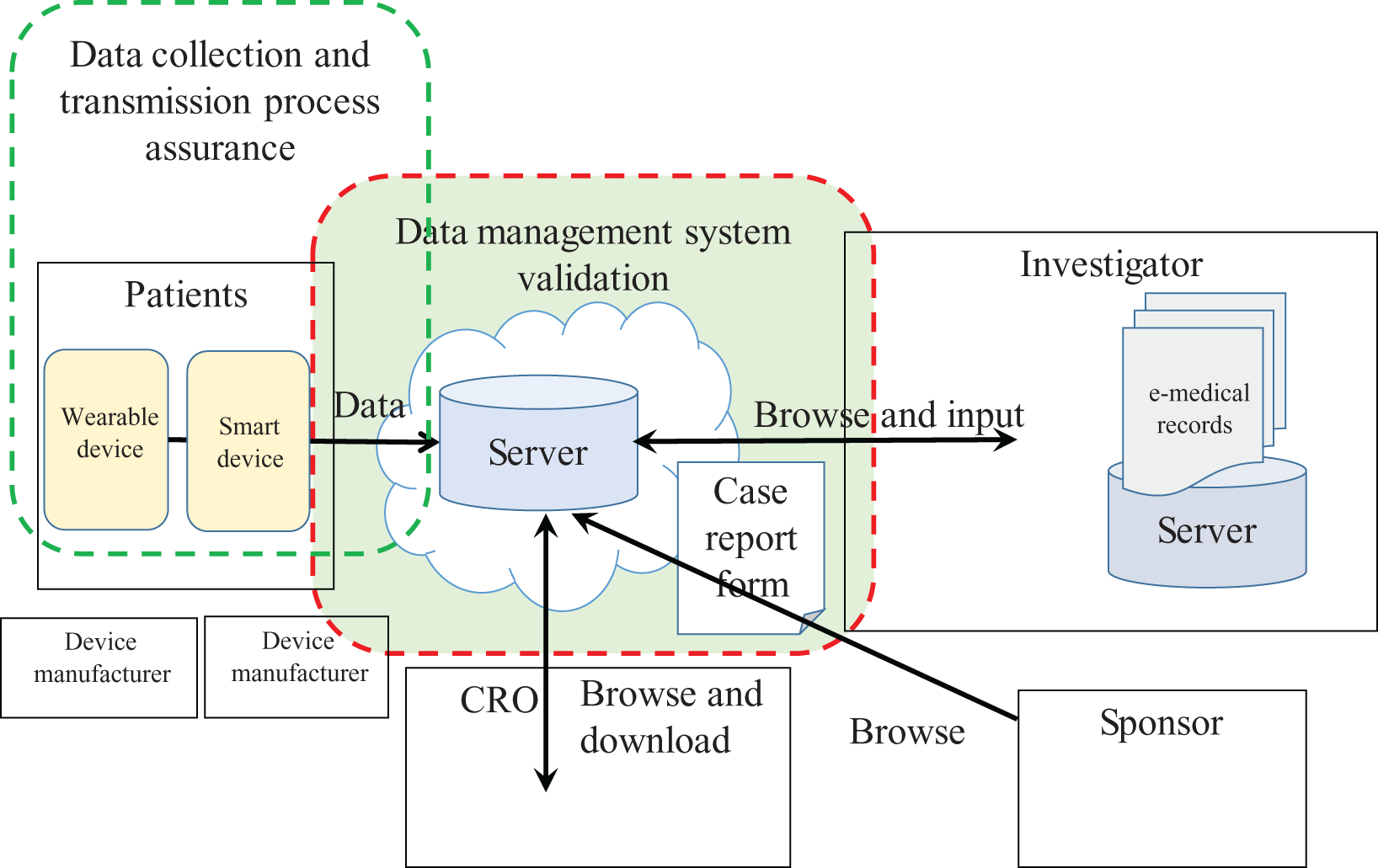
Computerized system to collect patient data with smart devices and wearable devices.
Patient data such as blood pressure, for example, is intermittently collected with a wearable device and transmitted from the wearable device to a smart device. The data is temporarily kept in the smart device and transmitted to the central server so that the data is archived in a secure environment.
The CRO ensures that the system is validated and works well just around the central server, because clinical programmers and data managers from the CRO are key players in the business processes and they know what functionalities are needed to do their job. They are also fundamental in activities involved in the validation of the computerized system. They might be involved in the analysis of user requirements, definition of functional specifications, review of functional design, and testing of user acceptance.
Key functions to be tested are as follows: trial set up with data entry screen definitions and clinical programming, data entry operation with univariate data checking, query handling with multivariate computer checking and resolution management, reporting, data storage, and so on (Table 2).
The sponsor should communicate with the CRO and receive relevant information about how the clinical data management system is validated by the CRO. The CRO might disclose relevant validation documents to the sponsor on an as-requested basis. In other words, when a sponsor outsources clinical data management to a CRO under contract, system validation is likely to be carried out by the CRO, instead of by the sponsor.
On the other hand, smart devices and wearable devices are COTS systems. Therefore, the CRO might have difficulty in validating and assuring the devices within the computerized data management system. It is assumed that devices have been assured and released by the device manufacturers in accordance with their quality management systems, even if they did not intend to conduct computerized system validation. It would be feasible for the CRO to gain an understanding of the product quality of the device by referring to the manufacturer’s quality management system.
Supplier Assessment
When a system owner intends to validate the system, official assessment of the suppliers related to the system should be conducted in a timely manner. Validation practitioners never complete computerized system validation without assessing suppliers. When a product has been developed and tested in the supplier’s factory, the supplier’s assessment is only one measure through which validation practitioners can know how the factory tests were conducted on the supplier’s side. Technical document verification is very meaningful and is fundamental to confirming that the necessary activities have taken place on the supplier’s side, even if they occurred many years ago.
When a CRO establishes a clinical data management system involving smart and/or wearable device and provides a regulated company (sponsor) with its operation support services, the CRO becomes the supplier and the device manufacturers are sub-suppliers to the sponsor.
Sub-supplier assessment is one of the supplier’s essential activities. 8 The CRO should manage device manufacturers following previously agreed on procedures such as periodical reporting (Figure 2). The sponsor may be able to receive relevant information from the CRO and know how the CRO controls its sub-suppliers.
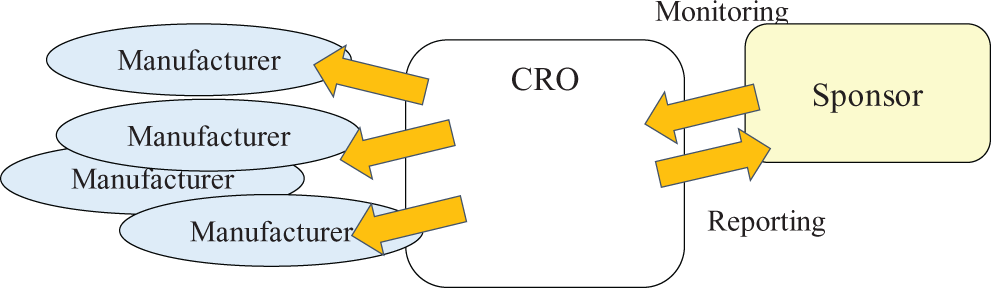
Sponsor interacts with contract research organization (CRO).
Computerized systems are constructed from products and/or applications developed and delivered by third-party suppliers. When a CRO supplies an electronic data management system involving smart and/or wearable devices, the CRO is supposed to have conducted a sub-supplier assessment with the device manufacturers. If this has not yet been done, the sponsor should ask the CRO to do so. Under “Supplier Good Practices” as defined in GAMP5, 8 assessment of sub-suppliers is one of the supplier’s key activities.
When a CRO intends to conduct sub-supplier assessment with a device manufacturer, a risk-based approach can be taken. Device risk evaluation might be done by focusing on its progressiveness, cumulative experience, number and location of end-users, and so on. We suggest rating the device risk “high” when the data management system is to deal with a pivotal trial. Considering limitations in financial and human resources, a postal audit is acceptable as a sub-supplier assessment for a system with low- or middle-level risk.
Risk-based Approach
Computerized systems are constructed from products and applications delivered by several suppliers and sub-suppliers. Although it is expected that suppliers assess sub-suppliers, suppliers might have difficulty assessing all sub-suppliers via on-site audits. Thus, the CRO (supplier) should take a risk-based approach by performing a postal audit unless the computerized system is intended to manage the clinical data of a pivotal trial. For a clinical data–handling system in a nonpivotal trial, the CRO may conduct a postal audit instead of an on-site audit (Figure 3).
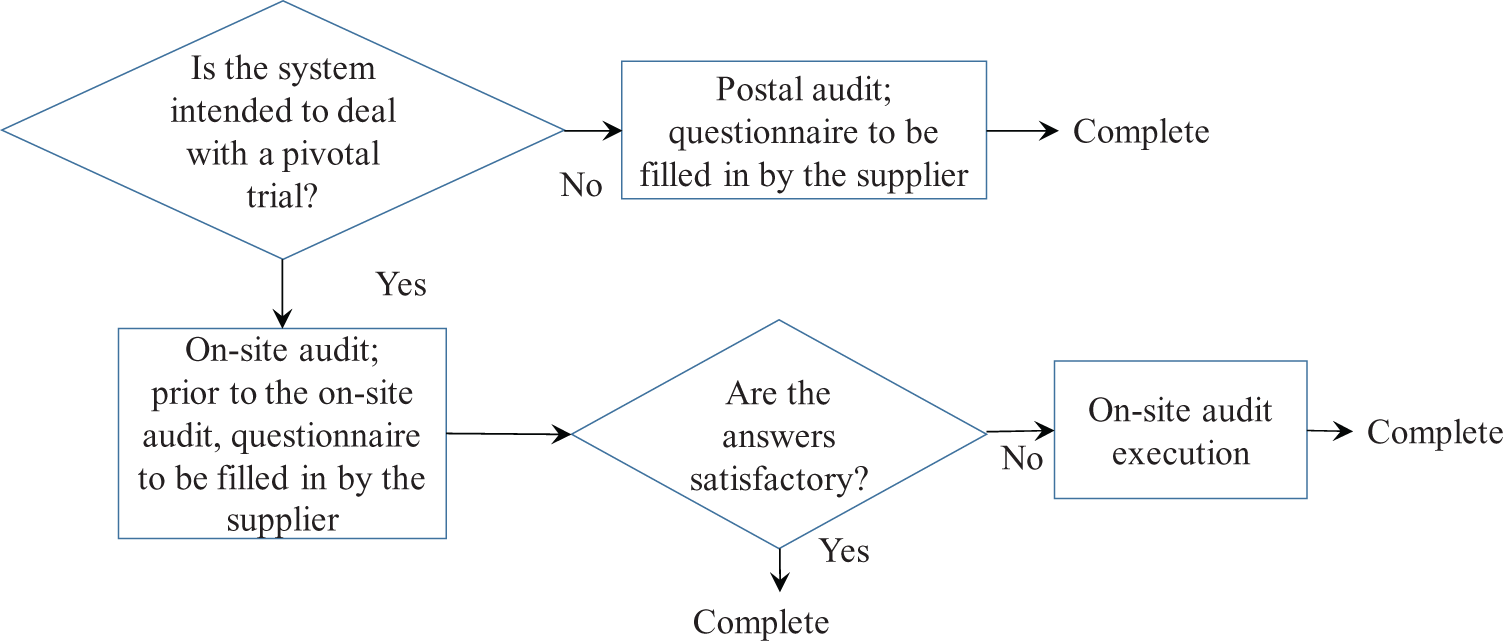
Risk-based approach to sub-supplier assessment.
It would be reasonable to take a risk-based approach to prioritize several supplier assessments. In general, supplier assessment is a time-consuming task requiring substantial financial and human resources.
Sponsors should have their own policies and standard procedures to conduct computerized system validation on clinical systems employed to evaluate efficacy of medicinal products in clinical trials. When a sponsor contracts out clinical data management operation to a CRO (supplier) with sufficient skill and technique to handle clinical data, the clinical data management system is likely to be validated by the CRO instead of the sponsor. The sponsor, however, is ultimately accountable for the system validation. The sponsor should make a service level agreement (SLA) with the CRO and ask the CRO to conduct system validation for the data management system in accordance with the sponsor’s own system validation policies and procedures.
In the case that the wearable device is marketed under the authorization of the health authorities, some technical documents might be available from the device manufacturer, or disclosed on the health authorities’ websites. In accordance with US federal regulations, medical devices are approved via the premarket approval (PMA) pathway or 510(k)-cleared as “substantially equivalent” to formerly approved devices in the United States. In Japan, the medical device database is available for obtaining information about marketed medical devices categorized by 4 levels. The sponsor and/or the CRO might take advantage of such information to make sure the device works well. Such confirmation could be a part of the validation activities.
Discussion
The sponsor is required to validate the clinical systems utilized to manage clinical data in accordance with its policies and standard procedures. For this purpose, the sponsor should have their own quality management system so that they can successfully carry out validation activities via an organized framework.
Electronic management systems involving smart and/or wearable devices are likely to be constructed with cloud computing technology, because recent technological progress is so fast and IT suppliers and CROs have lots of professionals and subject matter experts with sufficient skill and experience to develop and maintain such systems appropriately and efficiently. It would be reasonable to set up an application server on the service provider side. Computerized system validation frameworks have been established with traditional systems, but validation of systems involving cloud computing services is a different story. 17 Involvement of suppliers depends on precisely what activities are outsourced. 18 The sponsor is responsible for making a service level agreement with the CRO so that the CRO conducts the necessary activities regarding system validation in accordance with the sponsor’s policies and standard procedures.
The sponsor should have its own policies and standard procedures with which to conduct computerized system validation on the clinical systems utilized to evaluate the efficacy of medicinal products in clinical trials. When the sponsor contracts out clinical data management to a CRO, the sponsor should ask the CRO to conduct system validation for the system in accordance with the sponsor’s own system validation policies and procedures.
When some of the clinical data are collected by means of smart and/or wearable devices, the device manufacturers (sub-suppliers) need to be assessed by the CRO. If assessment has not yet taken place, the sponsor should ask the CRO to conduct sub-supplier assessment with the relevant device manufacturers.
When the CRO intends to conduct sub-supplier assessment of a device manufacturer, a risk-based approach can be taken. A postal audit with a questionnaire to be filled in by the manufacturer is one acceptable measure.
The research presented here has some limitations. First, although we designed a general model of a data management system involving smart and/or wearable devices, the processes of data collection might vary greatly according to therapeutic area involved, disease severity, and patient condition. Second, the approach to validation of cloud computing systems depends on how deeply the cloud computing service supplier is involved in establishing, verifying, and/or operating the system. Additionally, when it comes to patient-reported outcomes via smart devices, the appropriateness of the outcome measure’s questions was not investigated, although this point impacts highly on clinical trial results.
Conclusion
Electronic data collection is a strong measure with which to clarify risks of diseases caused by environmental triggers, genetic factors, and so on, as targeted in the Precision Medicine Initiative Cohort Program. A special interest group consisting of experts from relevant industries and academia discussed and designed a general model of a data management system involving smart and/or wearable devices in clinical trials and indicated points to consider in its system validation.
It is still unclear how to validate and qualify computerized systems to collect and/or manage electronic clinical data, because this matter has not yet been sufficiently discussed. We hope that this research paper helps validation practitioners of electronic data management systems utilized in clinical trials. We expect that this framework can be versatilely applied to investigator-initiated trials and nonregistration trials as well as to sponsor-initiated registration trials.
Footnotes
Author Note
This research was presented at the GAMP Seminar, October 23, 2015, Tokyo, Japan.
Declaration of Conflicting Interests
No potential conflicts were declared.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by JSPS KAKENHI Grant Number JP26560270.