
Abstract
Background:
Recently, numerous pharmaceutical sponsors have expressed a great deal of interest in the development of biosimilars, which requires clinical trials to demonstrate that the pharmacokinetic (PK) and clinical efficacy are equivalent. Pharmacodynamics (PD) may be used in evaluating efficacy if there are relevant PD markers available. However, in their absence, it is necessary to design the associated clinical trials to include efficacy measures as the primary endpoint.
Methods:
In this study, we propose a novel adaptive seamless PK and efficacy design with an efficient framework to remedy the risk of misspecification of efficacy parameters and to discontinue the trial evaluating the efficacy for futility based on the PK evaluation. Here, we consider the clinical development of biosimilars including their evaluation in patients rather than healthy volunteers under a situation where both PK and efficacy parameters are required to demonstrate equivalence. The original idea of the proposed method was to organize a clinical trial that includes the statistical analysis of PK as an interim analysis, with sample size recalculation of the efficacy data.
Results:
Our simulation study indicated that the proposed design would allow trials to be more efficient than with the classical design.
Conclusions:
This proposal provides appealing advantages, such as a shorter time period, additional cost savings, and a smaller number of patients required.
Keywords
Introduction
Adaptive seamless designs are rapidly becoming more attractive to numerous pharmaceutical sponsors in every phase of the clinical trial process, including adaptive seamless phase I/II or adaptive seamless phase II/III trials. In the context of confirmatory clinical trials, numerous statisticians interested in these methodologies have focused on superiority trials. 1 –3 However, other types of trials, such as non-inferiority and equivalence between test and reference products, have also been attractive to the pharmaceutical industry. In particular, equivalence trials, called bioequivalence trials, are applied in the development of generic products. To confirm bioequivalence, pharmacokinetic (PK) parameters, such as area under the concentration-time curve (AUC) and maximum serum concentration (Cmax), are typically evaluated in healthy volunteers using two one-sided tests (TOST) 4 with log-transformed values. Although some methods, such as the adaptive sample size recalculation, have been proposed for bioequivalence trials, 5 –8 these are rarely used in the actual clinical setting. Because such trials are conducted by recruiting generally between 20 and 50 volunteers, inclusion of interim analyses in the trials is usually not reasonable.
While numerous pharmaceutical sponsors have expressed interest in bioequivalence trials, considerable interest in developing biosimilars is also growing. 9 There is no unified definition for biosimilars; however, according to the guideline of European Medicines Agency, a biosimilar is defined as a biological medicinal product that contains an active substance similar to that of the original previously authorized biological medicinal product. 10 Biosimilars differ from generic chemical products, for example, with respect to the complexity and heterogeneity of the molecular structure. 9,11 A reduction in healthcare costs for patients can be expected if a biosimilar is approved by regulators and placed on the market. However, characteristically, a larger number of subjects would be required to investigate and clinically develop a biosimilar than that is required for the development of a generic product because there are regulatory requirements that encourage sponsors to provide pharmacodynamic (PD) or efficacy data in addition to PK data. 10,12 That is, there must be no clinically meaningful difference between the test and reference products. Moreover, as far as the PK data goes, a PK trial is sometimes conducted using a parallel-group clinical trial design instead of a crossover design owing to a high risk of immunogenicity. 10 In this instance, a large number of patients would be required to declare PK equivalence. For instance, a trial that was primarily aimed at confirming the PK equivalence was conducted with 250 recruited patients based on the assumption that the coefficient of variation (CV) was 50%. 13
In anticipation of the impending expiration of a number of patents for biological medicinal products in numerous countries, some statistical methods have been developed for evaluating biosimilarity. Methods for assessing biosimilarity with respect to variability between the test and reference products have been investigated. 14 –18 As a biosimilarity measure, a biosimilar index based on the concept of reproducibility probability has been proposed and discussed. 19 –21 Chiu et al 22 discussed the use of a Bayesian method that uses prior information. Pan et al 23 proposed a Bayesian group sequential design that incorporates information adaptively using a calibrated power prior. Chow et al 24 proposed methods for assessing biosimilarity based on the assumption that a biomarker is predictive of the clinical outcome. Li et al 25 proposed a biosimilarity trial design for evaluating clinical efficacy with asymmetrical margins. Liao and Darken 26 developed a method for assessing biosimilarity by comparability of critical quality attributes. Furthermore, a three-arm parallel design, which consists of one test and two reference products from two different batches, was provided to investigate biosimilarity. 27 When the three-arm parallel design is employed, the approach with the use of the frequency estimator criterion was also proposed to assess biosimilarity. 28 In summary, most methods currently focus on one specified trial. Therefore, little methodology that enables the performance of multiple trials seamlessly has been developed.
Here, two main trials served as motivating examples for confirming PK and efficacy in establishing the equivalence of a biosimilar of the innovator infliximab (Remicade). 13,29 These trials indicated that a clinical trial to establish the efficacy equivalence is often required in case any relevant PD markers are unavailable and that a PK trial could be conducted in patients rather than healthy volunteers. Although these trials were conducted separately, they each had a primary endpoint that was intrinsically set to determine PK and efficacy equivalence, respectively.
Based on this motivating example, in this study, we considered the clinical development of biosimilars with an emphasis on the necessity of demonstrating equivalence between the test and reference products by including both PK and efficacy as primary endpoints. We assumed that patients with the same disease conditions were targeted to provide equivalence data for the PK and efficacy. Methods using adaptive seamless designs, which allow sample size recalculation based on interim data, were applied in this setting. The adaptive seamless PK and efficacy design, which incorporates trials to establish both PK and efficacy equivalence, allows trials to be more efficient than classical trial designs.
This paper is structured as follows. First, we introduce a motivating example for the development of one biosimilar. Next, we propose a novel adaptive seamless PK and efficacy design and then lay the framework of our proposed design in the section “Methods.” The section “Simulation Study” presents a simulation study. Finally, in the section “Discussion and Conclusions,” we conclude the paper with a discussion.
Motivating Example
First, we introduce a motivating example for the development of a biosimilar to the innovator infliximab (Remicade), which is a monoclonal antibody against tumor necrosis factor-alpha that is used to treat patients with active rheumatoid arthritis who have shown an inadequate response to methotrexate. 30 However, a major hurdle for patient access is the high medication cost. Therefore, there is a lot of interest in developing a biosimilar of the innovator monoclonal antibody. Biosimilars of infliximab (Inflectra and Remsima) were recently launched in several markets across numerous countries. 31 During the development of Remsima, two main clinical trials were performed. The trials were PK and phase III studies that demonstrated equivalence using PK and efficacy endpoints, respectively. 13,29 Both trials were designed as randomized, double-blind, parallel-group clinical trials, and their primary endpoints were the AUC and Cmax for the PK trial and the American College of Rheumatology 20% (ACR20) improvement 32 for the efficacy trial.
For the PK study, the required sample size was calculated to be 196 based on the equivalence margin of 80% to 125%, power of 90%, and TOST with a significance level of 5% under the assumption of a geometric mean ratio of 1.00 and a CV of 50%. In addition to PK, efficacy and safety were also compared in this study. In contrast, the sample size of the phase III study was 468, which was the sample size required to achieve 80% power to meet the equivalence margin within ±15% for ACR20 at a specific time point under the TOST with a significance level of 2.5%, assuming an expected response rate of 50% in both groups. As secondary endpoints, additional efficacy, immunogenicity, safety, PK, and PD were assessed; however, no adjustments for multiplicity were performed.
Consequently, equivalence for the primary PK and efficacy endpoints was established in each trial. Each confidence interval (CI) was in the range of the corresponding equivalence margins. For the PK parameters, the geometric mean ratios (90% CI) were 1.05 (0.94-1.16) and 1.02 (0.95-1.09) for the AUC and Cmax, respectively. As an efficacy parameter, the ACR20 response rates for each group were 60.9% and 58.6%, and the difference in the response rates (95% CI) was 2% (−6% to 10%) for the intention-to-treat population, whereas the ACR20 response rates were 73.4% and 69.7%, with a difference of 4% (−4% to 12%) for the per-protocol population.
Note that each between–product group difference observed varied from the prespecified settings. In confirmatory clinical trials, misspecifications often occur regardless of careful planning. For example, the observed geometric mean ratio of the AUC deviated from the prespecified value of 1.00, whereas the observed response rates for each group in the per-protocol population differed greatly from the prespecified value of 50%. Even if a sponsor in one country designs a trial based on trials conducted in other countries, the observed values from that trial will not necessarily be consistent with those of the other countries owing to reasons such as possible race- or measurement technique–related differences between countries. 29,33 These misspecifications have been accounted for by recruiting more patients, which allows for more drop-out or exclusion of population sets than required. In addition, the observed response rates for each group differentially deviated from the expected value of 50%, and this was a conservative setting because the range of the CIs tends to widen. Because of these remedies, the targeted sample sizes for both trials were in effect set to approximately 1.15 to 1.20 times the required sample size. Instead, to compensate for this risk of the failure to demonstrate equivalence, a sponsor could consider increasing the sample size in midcourse, that is, sample size recalculation within adaptive seamless design.
Methods
In this section, we consider the clinical development of biosimilars under a randomized parallel-group design with two arms, test (T) and reference (R) products. Similar to the motivating examples of the Remsima, 13,29 we assume that a crossover design is not a feasible option for the trial design because of the associated problem of carryover effects, although crossover designs are often used in the development of biosimilars where recruitment for the study population is targeted at healthy volunteers. 34,35 Based on the motivating example involving two trials, a single trial is conducted for the evaluations of both the PK and efficacy, and their confirmations constitute the interim and final analyses, respectively. To compensate for the misspecification of parameters at the planning stage, an interim analysis will be performed for the final efficacy confirmation that requires more patients than the PK confirmation. Figure 1 shows the proposed framework of the adaptive seamless PK and efficacy design. Note that the objective of the trial is to determine the equivalence of both PK and efficacy.
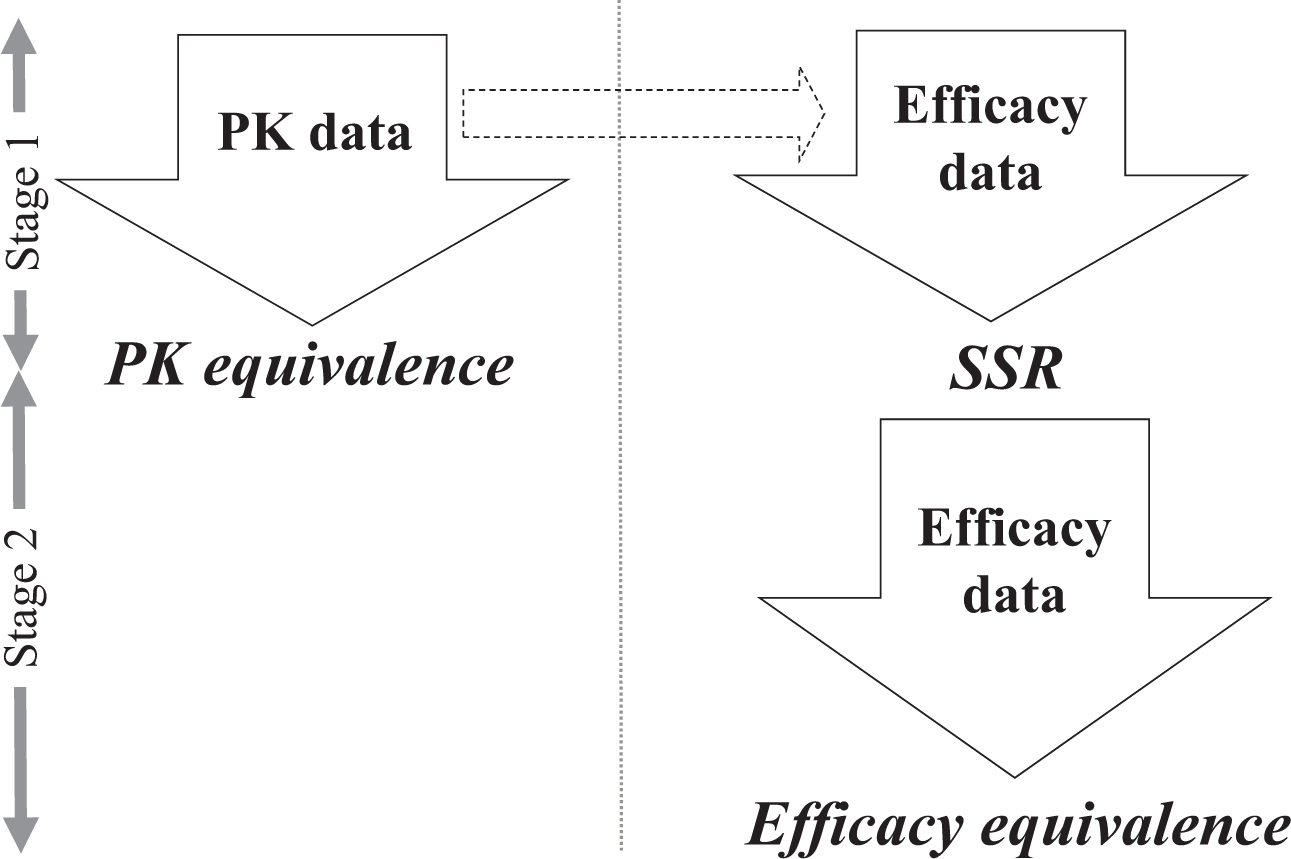
Proposed adaptive seamless pharmacokinetic (PK) and efficacy design. SSR, sample size recalculation.
Collecting Both PK and Efficacy Data From the First Stage
Here, we considered that not only PK data but also efficacy data could be obtained from the PK trial. To achieve this, we assumed that both the PK and efficacy trials should be conducted with patients who have a common disease and are receiving similar dosage regimens.
The strength of this study is that it constitutes a clinical trial that includes the statistical analysis of PK as an interim analysis of the efficacy data. Therefore, we consider a two-stage design based on the assumption that both the PK data and efficacy data are obtainable from the first stage, as described in Figure 1. The data from the first stage can subsequently be used to test the PK equivalence and arrive at an interim decision for the efficacy equivalence, whereas only the efficacy data are obtainable from the second stage to test the efficacy equivalence.
Sample Size Adjustment for Efficacy Endpoint of Interim Analysis
The interim analysis conducted in an unblinded fashion allows for the option to recalculate sample size for the subsequent stage if the interim result indicates a potential benefit for sample size recalculation. Note that the interim decision for the subsequent stage is only based on the efficacy data of the interim analysis.
Let Z1 and Z2 denote the test statistics until the interim and final analysis, respectively. Without any interim analysis from the first-stage data, the hypothesis testing would be conducted conventionally, with
Mehta and Pocock
36
have proposed methods in which the sample size is increased if the interim results are promising and the decision is made based on the conditional power. Recently, a thorough investigation of this approach has been conducted and reported in several articles in the context of a superiority trial.
37,38
In this study, we consider this approach in evaluating the equivalence of an efficacy endpoint. The conditional power given by
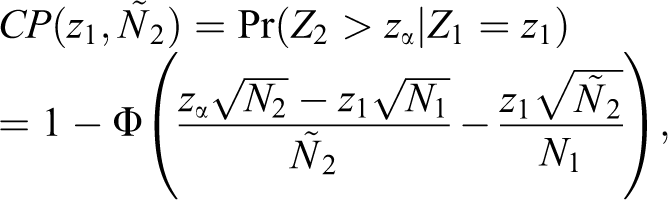
where N1 and N2 are the planned sample sizes for the interim and final analysis, respectively, and
The promising zone approach arrives at the interim decision by defining the region that represents the promising zone as follows:
Case 1 (Promising): Case 2 (Otherwise, ie, favorable or unfavorable): → Continue to the planned N2,
where cpmin is the prespecified lower probability of the conditional power. If the interim conditional power is deemed promising, the sample size is increased to
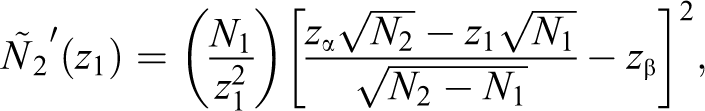
with the restriction that the maximum sample size increase is Nmax. Thereafter, the critical value for the final analysis, in exchange for zα, can be adjusted to
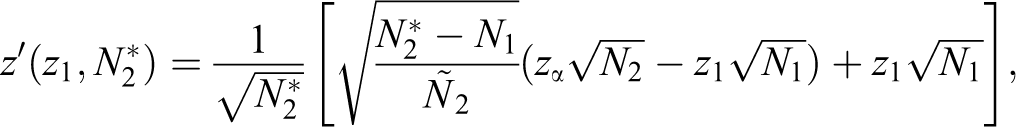
which holds that
The final analysis would also be conducted using
With an equivalence trial, the interim result, z1, for efficacy is constructed using the following null hypotheses for the efficacy endpoint:
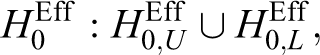
that is,
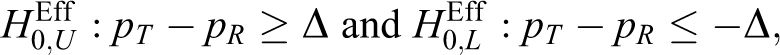
where pg is the response rate for the group
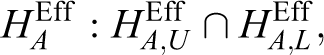
that is,
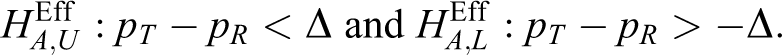
PK Equivalence Including Remedy for Insufficient Sample Size
We shall begin this section by describing the hypotheses for the PK evaluation. For the PK equivalence, the null hypotheses for PK are constructed as follows:
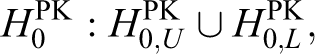
that is,
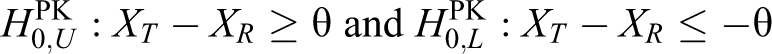
where Xg is the log-transformed mean of the PK parameters such as the AUC or Cmax for group
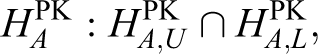
that is,
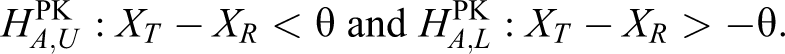
In the bioequivalence trial, the θ is traditionally set as the log-transformed 1.25, which corresponds to a range of 80% to 125% under the
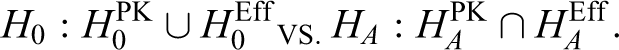
Note that the H0 is rejected only if the equivalence for both the PK and efficacy is established. This meets the requirement based on several guidelines in which both the PK and efficacy are required to show equivalence when developing biosimilars. 10,12
Figure 2 shows the detailed framework used to declare the equivalence of both the PK and efficacy within the adaptive seamless PK and efficacy design as described in Figure 1. With respect to controlling the type I error rate for PK and efficacy equivalence under each significance level, a fixed sequence testing procedure
43
is incorporated into this framework. That is, the efficacy equivalence is tested only if the PK equivalence is declared. As shown in Figure 1, it organizes a clinical trial that includes the statistical analysis of PK as an interim analysis of the efficacy data because we considered that both PK data and efficacy data could be obtained from the sample size N1 in the PK trial. If PK equivalence fails to be declared, subsequent collection of efficacy data is discontinued for futility. On the other hand, collecting efficacy data is subsequently continued if PK equivalence is declared. Thereafter, sample size recalculation from N2 to
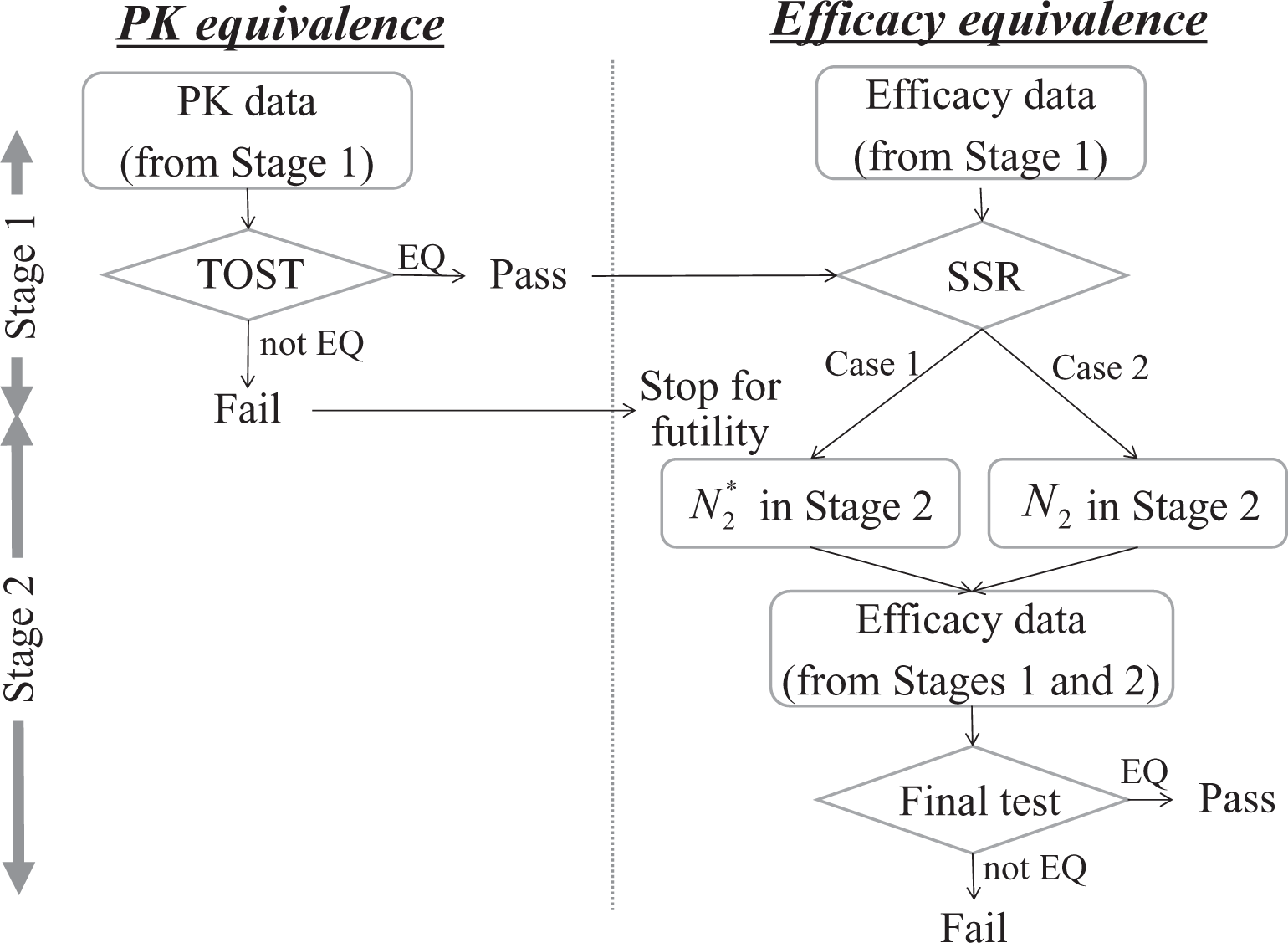
Framework that determines equivalence of both pharmacokinetic (PK) and efficacy. “EQ” and “not EQ” denote where equivalence is declared and not declared, respectively. SSR, sample size recalculation; TOST, two one-sided tests.
Simulation Study
Here, we evaluated the efficiency of the adaptive seamless design proposed in this study. This is to enable the presentation of their operating characteristics based on the design parameters such as the geometric mean ratio and CV in the PK trial, and response rates for each group in the efficacy trial. The assumptions made for the PK and efficacy trial designs and the simulation setting are presented in the subsection “Simulation Setting.” In the subsection “Simulation Results,” we demonstrate the effectiveness of the proposed adaptive seamless PK and efficacy design based on the power and expected sample size.
Simulation Setting
First, we consider a randomized, parallel-group clinical trial with two arms, which are the test (T) and reference (R) product arms. We assume that E is set as the biosimilar, and the sample size recalculation for the efficacy endpoint under the adaptive seamless PK and efficacy design as described in the section “Methods” is conducted with an interim analysis, which plays a role in the statistical analysis of the PK data.
Based on the examples,
13,29
we suppose that the overall planned sample size is set as
To assess the sample size recalculation for efficacy equivalence, the lower probability of the conditional power cpmin is set at 50% or 33%.
36,44
We further assume that the magnitude of the sample size increase is set as
For reference, the power and expected sample size are also illustrated when a fixed design is used in exchange for the adaptive seamless PK and efficacy design. Regarding the expected sample size using the fixed design, we consider that the PK and efficacy trials are conducted separately, suggesting that the efficacy data are not available for the PK confirmation. That is, the PK trial is assumed to be conducted and the efficacy trial is subsequently conducted separately under the fixed design. Hence, if the PK equivalence fails to be declared, the efficacy trial is not implemented.
Simulation Results
We evaluated the efficiency of the adaptive seamless PK and efficacy design proposed based on the type I error rate, power, and expected sample size. The calculation was based on 500,000 simulations with respect to the type I error rate and 10,000 with respect to power and expected sample size to display the operating characteristics.
Table 1 shows the probabilities of rejecting the null hypotheses
Type I Error Rates.a
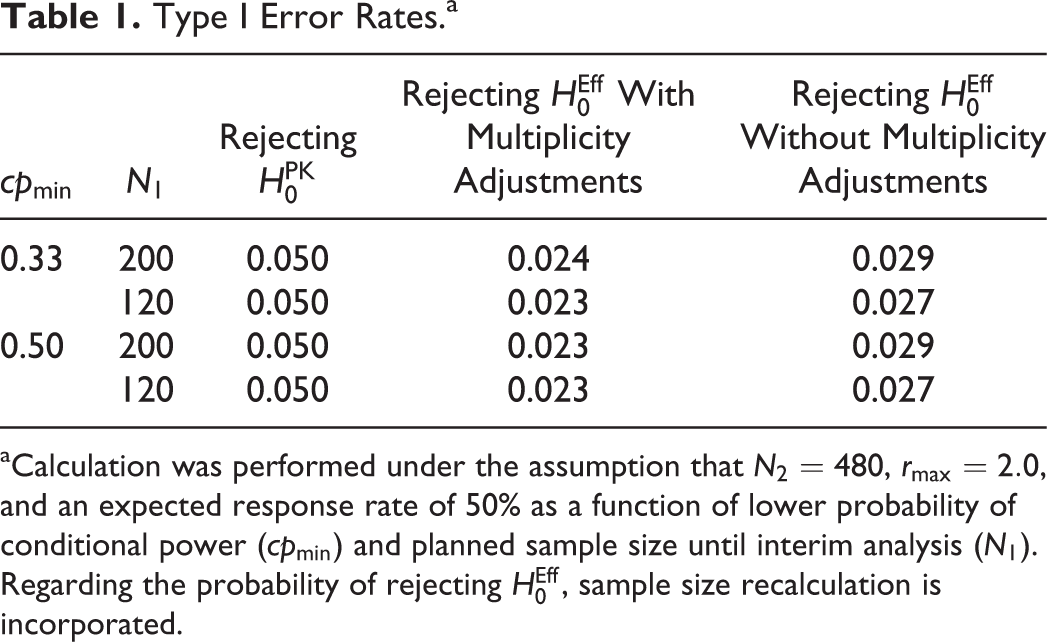
aCalculation was performed under the assumption that
The comparison of powers between the fixed design and the adaptive seamless PK and efficacy design in the final analysis is shown in Table 2. These powers are defined as the probability of rejecting
Powers of Adaptive Seamless Pharmacokinetic (PK) and Efficacy and Fixed Designs.a
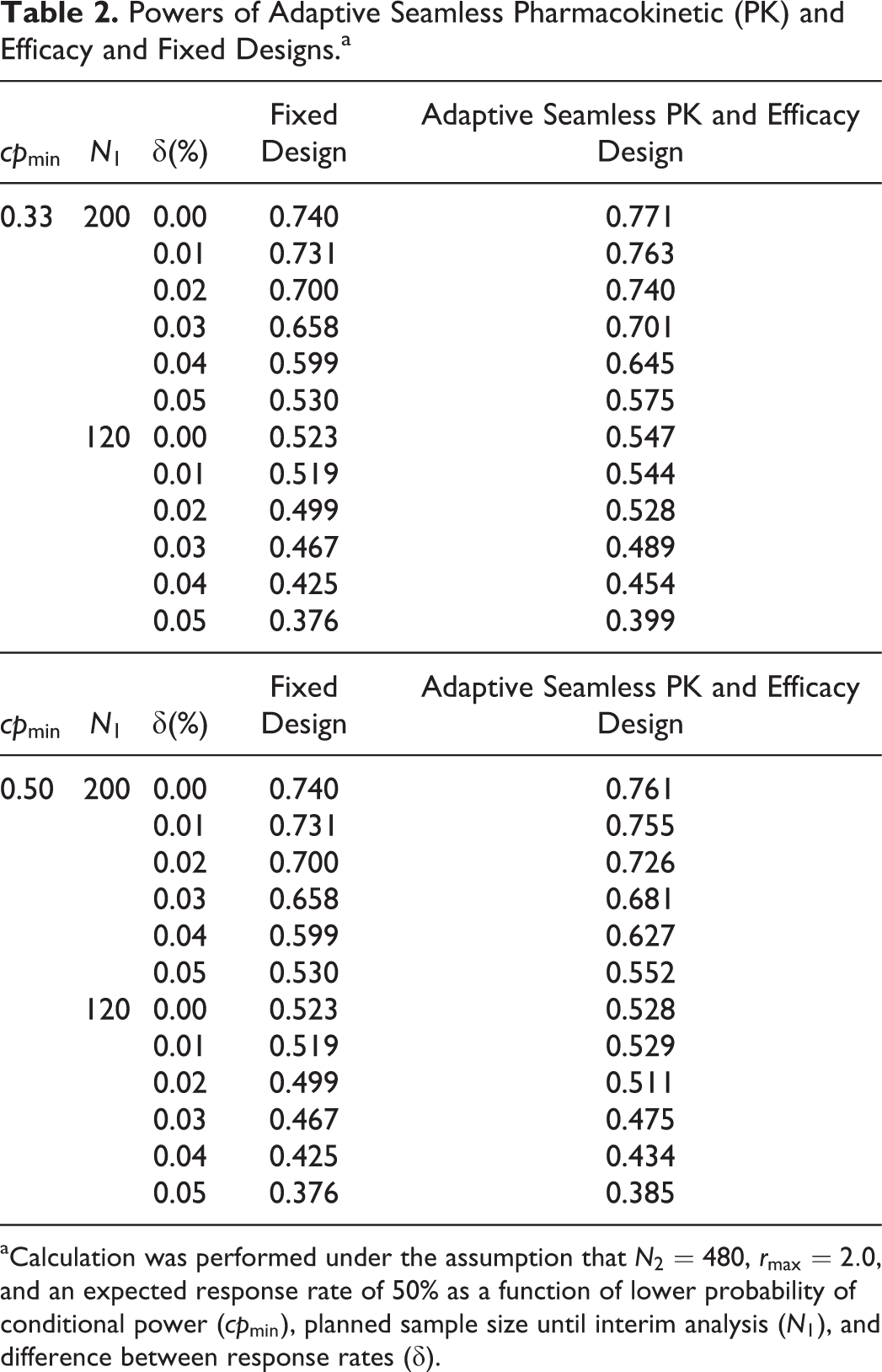
aCalculation was performed under the assumption that
We showed that the power is improved by the use of the frameworks for PK and the sample size recalculation in the promising zone approach for the efficacy of the adaptive seamless PK and efficacy design. The gain of power is attributable to the increase in sample size. Table 3 shows the expected sample size for achieving the corresponding power illustrated in Table 2. Note that the expected sample sizes for the fixed design do not exactly correspond to the sum of the interim and final sample size (that is, N1 and N2) because of the failure to declare PK equivalence in the PK trial preceding the efficacy trial. It is obvious that the approach that uses the adaptive seamless PK and efficacy design is more efficient than the approach that uses the fixed design, where N1 is set at 200 and the expected sample size is decreased because the evaluations for both the PK and efficacy are conducted separately in the latter design. Hence, it is necessary to set a sample size that has sufficient power. Furthermore, the increase in power and expected sample size is higher when cpmin is set at a lower value.
Expected Sample Sizes Corresponding to Powers in Adaptive Seamless Pharmacokinetic (PK) and Efficacy and Fixed Designs.a
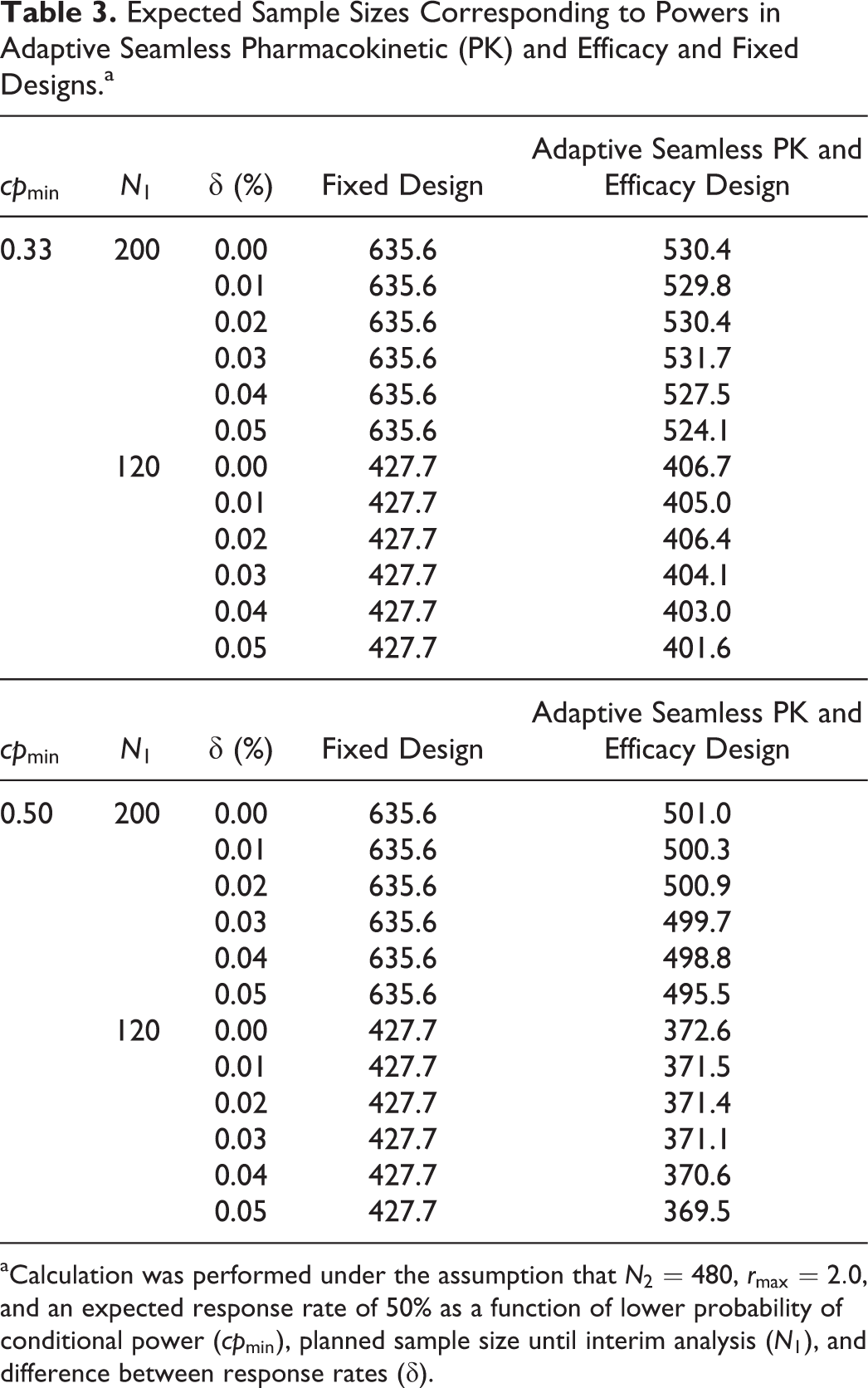
aCalculation was performed under the assumption that
Discussion and Conclusions
The aim of this study was to propose a novel adaptive seamless PK and efficacy design for establishing the equivalence of both PK and efficacy in clinical trial phases for the development of biosimilars. The proposed design, which allows sponsors to develop biosimilars over shorter time periods, leads to additional cost savings, requires a fewer number of patients for trials, and would be an appealing strategy for the efficient implementation of clinical trials. This enhanced process would consequently accelerate product approval by regulatory agencies. Note that there is still controversy about statistical analysis for biosimilar development, even though related guidelines 10,12 have been issued from the regulatory agencies. For instance, choosing the margin, primary endpoint, and primary time point for efficacy represent the issues and challenges with respect to biosimilar development. Hence, consultation with regulatory agencies must be required before applying the proposed design, which has originality specific to biosimilar development and offers benefits even considering the issues and challenges.
Our proposed framework is an attractive option with respect to the total trial period as mentioned in the subsection “PK Equivalence Including Remedy for Insufficient Sample Size.” For efficacy equivalence, sample size recalculation was incorporated in the adaptive seamless PK and efficacy design to compensate for the risk of misspecification of the efficacy parameters. The study is subject to early termination in the efficacy part if PK equivalence fails to be declared. The power was improved as shown in Table 2, but not dramatically increased with sample size recalculation for the efficacy part because we considered that the preplanned sample size was adequate to achieve the target-level power of 80%. Note that the trial should be planned carefully to estimate the sample size and should not be set up deliberately with an underestimated sample size with insufficient power solely dependent on the sample size recalculation. With the planned sample size with insufficient power, the expected sample size using the adaptive seamless PK and efficacy design is larger than that using a fixed design; however, it is smaller when a sample size with sufficient power is used. The promising zone approach also enables the trial to avoid implementing further support when the interim result deems it obviously unpromising. This would reallocate and optimize the additional investment of resources. However, a downside of sample size recalculation is that the statisticians associated with the sponsor can grasp the interim conditional power based on the additional sample size to be enrolled during the subsequent stage. For the interim analysis, sample size recalculation should be conducted with strict adherence to the operating procedures, which are incorporated in the charter for the interim analysis while setting up an independent data monitoring committee. This step would prevent operational bias and maintain the trial integrity, even if the trial is an equivalence design, as the US Food and Drug Administration has cautioned. 45
It is noteworthy to mention that this work was limited to a specific situation where there are no relevant PD markers for measuring the efficacy in clinical trials. In addition, we propose the adaptive seamless PK and efficacy design with the restriction that both PK and efficacy trials are required to be conducted with patients. This is because the premise of this study was based on the characteristics of biosimilar development trials that are often conducted in patients rather than in healthy volunteers. Although healthy volunteers are used in most applications for biosimilars, 35 the development of biosimilars has a greater possibility of targeting patients hereafter owing to the high molecular complexity of biosimilars. For example, the biosimilar of trastuzumab (Herceptin), which is similar to the biosimilar infliximab because it is a monoclonal antibody, has been developed. 46 The proposed adaptive seamless PK and efficacy design would also be applicable in the development of the biosimilar trastuzumab because PK and phase III studies 47,48 have been conducted for patients with anti–human epidermal growth factor receptor 2 (HER2)–positive metastatic breast cancer to demonstrate equivalence for PK and efficacy endpoints, respectively. In addition, there would be a controversial issue with respect to the assumption that patients who have common diseases and are being treated with similar dosage regimens are targeted to provide equivalence data for PK and efficacy. Note that in the motivating example described in the subsection “Motivating Example,” the targeted patients for the PK study had ankylosing spondylitis, 13 whereas those for the efficacy study had rheumatoid arthritis. 29 However, there is also an example in which targeted patients were rheumatoid arthritis even in the PK study. 33 Moreover, our framework was constructed using parallel-group clinical trial designs based on the two main trials performed in the development of Remsima. 13,29 Because crossover designs for PK trials are often used, the clinical trials that consist of PK evaluations using crossover designs and efficacy trials using parallel-group designs could be extended for use with the adaptive seamless PK and efficacy design. In this case, the adaptive sequential design used for PK confirmation 5–8 would be an additional option for the PK trial. As a further and practical consideration, a multiple testing issue for multiple PK endpoints would be needed in addition to the fixed sequence testing procedure considered between PK and efficacy endpoints because two PK endpoints, that is, AUC and Cmax, are often evaluated in practice in the PK trial. 49 In addition, several types of AUC are often set as primary endpoints. For instance, AUCs from time zero to predicted infinity and from time zero to the last measurable concentration were assessed in addition to Cmax as primary endpoints in the PK study within the development of the biosimilar adalimumab. 50 Further, other PK parameters, such as tmax, volume of distribution, and half-life, should be set as secondary PK endpoints, whereas AUC and Cmax are frequently set as primary PK endpoints. 10 In the motivating PK trial, 13 nine parameters were set as secondary endpoints, whereas AUC and Cmax were set as primary endpoints. Although multiplicity for secondary PK endpoints was not usually addressed and only primary PK endpoints were needed to demonstrate equivalence statistically, providing these secondary PK parameters is necessary to conclude biosimilarity in practice.
In conclusion, our study proposed a novel method for developing biosimilars using an adaptive seamless design that enables sample size recalculation based on interim data and incorporates trials to establish both PK and efficacy equivalence. Furthermore, the newly proposed design allows clinical trials to be more efficiently conducted than conventionally designed methods, thereby reducing costs, saving time, and providing an attractive option for pharmaceutical sponsors.
Footnotes
Acknowledgments
The authors thank the two reviewers for their helpful comments that greatly improved the paper.
Conference Presentation
This work was partially presented at the XXVIIIth International Biometric Conference in Victoria, Canada, July 10-15, 2016.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.