
Abstract
Background:
In this study we compared Swissmedic’s (SMC’s) regulatory marketing authorization decisions to those of the US Food and Drug Administration (FDA) and European drug regulatory authorities (EU). We investigated the overall similarity of the regulatory decisions, approval, and postmarketing withdrawal rates in the 3 jurisdictions. In case regulatory decisions diverged, we analyzed the reasons for rejection of marketing authorization applications (MAAs).
Methods:
The study comprises 255 new molecular entity (NME) MAAs assessed by SMC by the EU and FDA between 2005 through 2014. Study parameters included the regulatory decision, postmarketing withdrawal rates, and the official reasons for rejection.
Results:
Regulatory decisions converged to a high degree among all 3 agencies (between 84% and 90%). SMC’s average approval rate (84%) was slightly lower than those of the FDA (87%) and the EU (91%). Postmarketing withdrawal rates were generally low (4%-5%) but were 3 to 5 times higher when decisions among the drug regulatory authorities (DRAs) diverged. SMC’s primary grounds for rejection were lack of efficacy (45%) and safety (40%).
Conclusions:
The 3 investigated DRAs adhere largely to the same scientific principles and regulatory guidelines; therefore, remaining disparities ought to be considered in a cultural, legal and public health priority context.
Background
In order to fulfill its legal responsibilities and ensure the timely access of safe and efficacious innovative new medicinal products to the Swiss market, Swissmedic (SMC) is obliged to continuously optimize and streamline its drug evaluation processes. SMC actively fosters relationships with a number of foreign drug regulatory authorities (DRAs) and endeavors co-operation in form of agreements on information exchange such as Memorandum of Understanding (eg, with Health Canada, the Australian Therapeutic Goods Administration and the Singaporean Health Sciences Authority), Confidentiality Commitment (eg, with the US Food and Drug Administration [FDA]) and Exchanges of Letters (eg, with the EU). 1 The sharing of scientific information and views on the interpretation of marketing authorization data accelerates patients’ access to innovative new medicinal products and saves resources due to reduced duplication of work. Although in recent years, many drug regulatory requirements have become increasingly harmonized on a global scale, divergence of regulatory decisions continues to be a matter of debate among patient organizations, the pharmaceutical industry, and regulators.
To this end, a comprehensive analysis of the degree of consensus on decisions among SMC, the EU, and FDA as well as the concerns substantiating rejections has been lacking. Our aim was thus to analyze marketing authorization applications (MAAs) containing new molecular entities (NMEs) with SMC decisions dating between 2005 and 2014 and to compare the review outcomes with the respective decisions of the FDA and those taken in the EU. Moreover, when decisions between SMC and either the EU or the FDA diverged, the main reasons associated with rejection were analyzed and categorized.
Methods
In order to compare the regulatory decisions on MAAs evaluated by SMC with those evaluated by the FDA and the European Medicines Agency (EMA, Centralized Procedure) as well as EU national DRAs (Decentralized Procedures and national decisions), we screened 298 NME MAAs that received their final decision by SMC between January 1, 2005, and December 31, 2014. Medicinal products for veterinary use, blood products, radiopharmaceuticals, vaccines, complementary medicines and conditional marketing authorizations were excluded from this study because the legal basis and the marketing authorization pathways for these products are less comparable.
We obtained Swissmedic data from internal databases that are not in the public domain. In order to avoid multiple counting of the same application (eg, multiple negative decisions followed by a final positive decision), only the last official SMC decision was considered in the analysis.
We retrieved EMA data from the EMA Website 2,3 ; entries past December 31, 2014, were excluded. SMC applications were matched with EMA applications using the medicine name, the active pharmaceutical ingredient, and the company name. In cases we failed to match SMC applications to EMA applications, we searched the Heads of Medicines Agencies website 4 for products authorized through the decentralized and mutual recognition procedure and the EU Community Register 5 for nationally authorized medicinal products.
We retrieved the FDA data from the Drugs@FDA website 6 and the website of the Center for Biologics Evaluation and Research. 7 The FDA does not publish assessment reports in case of a negative outcome and in these cases it remains unknown to the public if a particular MAA has actually been reviewed or not. Unless the sponsor withdraws its MAA, it remains open until further data in support of an approval are submitted. 8 For this reason, in a number of cases we were unable to determine if an MAA was “rejected” by the FDA or whether it had never been reviewed.
SMC disposes of information regarding FDA applications because its marketing authorization applicants are required to indicate their product’s marketing authorization status at foreign regulatory authorities in Common Technical Document (CTD) module 1. For the purpose of this study, we considered MAAs to be rejected by the FDA if the medicinal product in question had not been approved by the FDA within 3 years after original filing of the dossier. This assumption is numerically supported by a study demonstrating that the median time between submission of an MAA to the FDA and the final decision is approximately 300 days and the review process of 90% of MAAs is completed in less than 800 days. 9
DRA decisions were classified as “positive” in case of approval of the MAA and as “negative” in case of a rejection of the MAA or a withdrawal by the applicant. In the EU, final decisions were issued by the European Commission in case of centralized procedures (CPs), by the reference member state (RMS) in case of decentralized procedure (DCP) / mutual recognition procedure (MRP), or by a national drug regulatory authority in case of national procedures.
In order to quantify and categorize the reasons for SMC rejections, we used preliminary (in case of applicant withdrawal) or official decisions letters and extracted the therein stated concerns substantiating the rejections. For MAAs with negative outcome at the EMA, we considered the European Public Assessment Reports section titled “What Were the CHMP’s Main Concerns That Led to the Refusal.” Statements substantiating SMC’s and EMA’s rejections were categorized according to a system originally established by the EMA. 10,11 We also applied the categorization system to determine a single “primary concern for rejection” for each selected MAA and considered all other concerns to be “secondary concerns for rejection.” If for a new drug clinically relevant efficacy could be demonstrated but there was concern about its clinical safety, we defined that clinical safety should be considered as the primary (decisive) reason for the negative outcome. FDA’s reasons for rejection could not be evaluated for the above-mentioned reasons.
Results
Comparison of Swissmedic, EU, and the FDA Decision Outcomes
Between 2005 and 2014, SMC made decisions on 298 NME MAAs that met the inclusion criteria. The annual number of SMC decisions ranged from 22 in 2005 to 42 in 2014 (Figure 1); in average, there were approximately 30 MAAs per year.
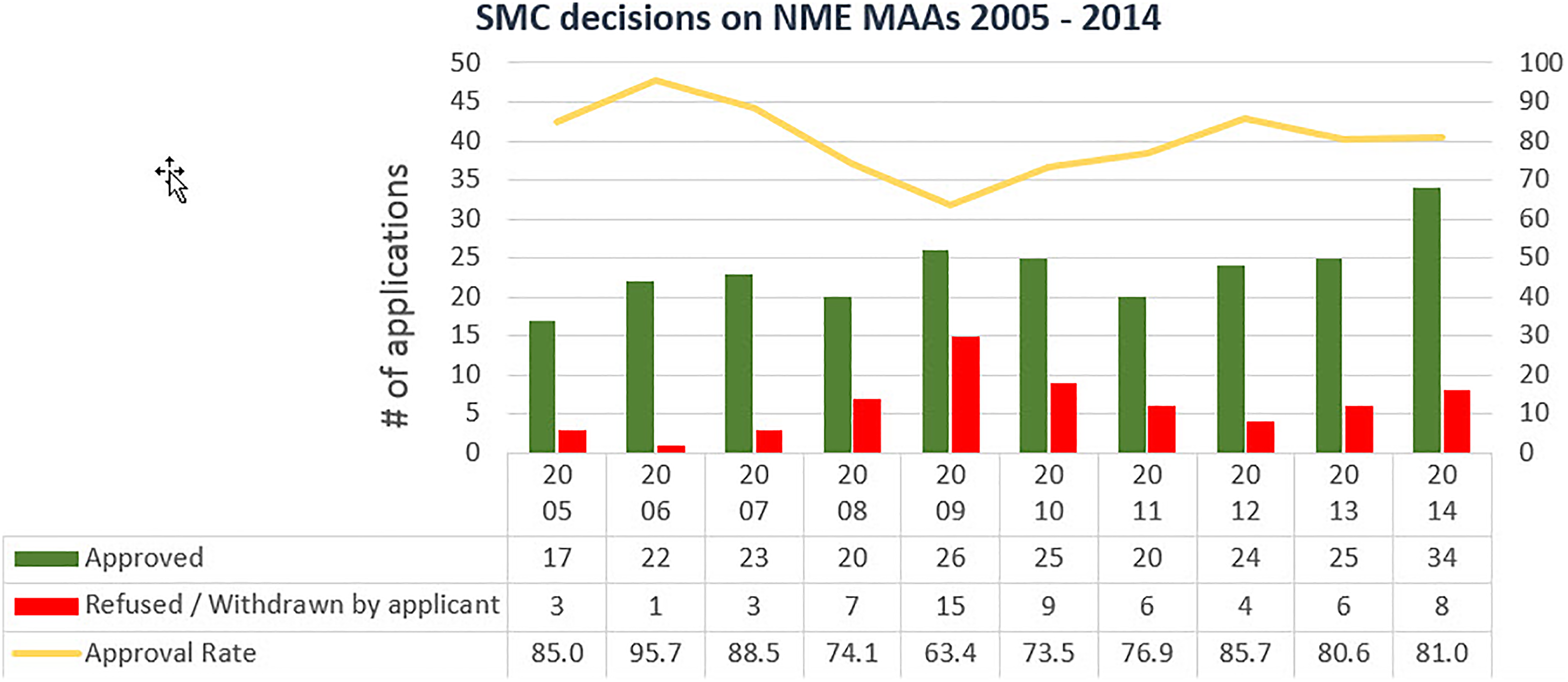
Number of positive decisions, negative decisions and approval rates of marketing authorizations applications completed between 2005 and 2014.
SMC’s approval rate of all these applications was 79%. Of the 62 (21%) MAAs rejected by SMC, 5 were refusals; in 57 cases, the applicants chose to withdraw the applications. Annually, approval rates ranged from 63.4% in 2009 to 95.7% in 2006.
For 255 of the 298 MAAs (86%) evaluated by SMC, the outcomes of EU’s and FDA’s assessment were known for this analysis. For the 43 remaining MAAs, no public information on the review status was available: for 39 MAAs it remained unknown whether they had ever been assessed by the FDA, for 6 MAAs no information about an EU assessment was available and for 2 MAAs both was the case. In the EU, 222 of the 255 NME MAAs were assessed by the EMA, 30 were reviewed through the DCP/MRP, and 3 were reviewed by at least 1 national DRA in the EU.
The 255 MAAs independently reviewed in all 3 jurisdictions allowed direct comparison of the DRA’s degree of decision consensus and the overall approval rates. The highest approval rate was reached in the EU (91%), the second highest in the USA (87%), and the lowest in Switzerland (84%) (Table 1).
Approval Rates and Number of Known/Unknown Decisions in 3 Jurisdictions.

Abbreviations: MAA, marketing authorization application; SMC, Swissmedic.
aNot reviewed / withdrawn by applicant before day 120 assessment milestone was reached.
bNot reviewed / rejected / withdrawn by applicant.
A total of 195 (76%) of the 255 MAAs were authorized in Switzerland, the EU, and in the USA, and 10 (4%) were not authorized in any of the 3 jurisdictions. The remaining 50 (20%) SMC decisions diverged from either the FDA’s or the EU’s.
In this cohort of 50 diverging decisions, in 19 cases (38%) the outcome was positive in Switzerland but negative in either the EU or the USA and in 32 cases (62%) the outcome was negative in Switzerland but positive in either of the 2 reference jurisdictions. In 15 cases (30%), Switzerland was the only country with a negative decision. There was not a single instance in which SMC was the only DRA granting a marketing authorization. However, the FDA made the most standalone negative decisions (16 cases, 32%). In the EU, only 3 (6%) standalone negative decisions were recorded over the 10 years (Table 2).
Distribution of Diverging Decisions on MAAs Reviewed in All 3 Jurisdiction.
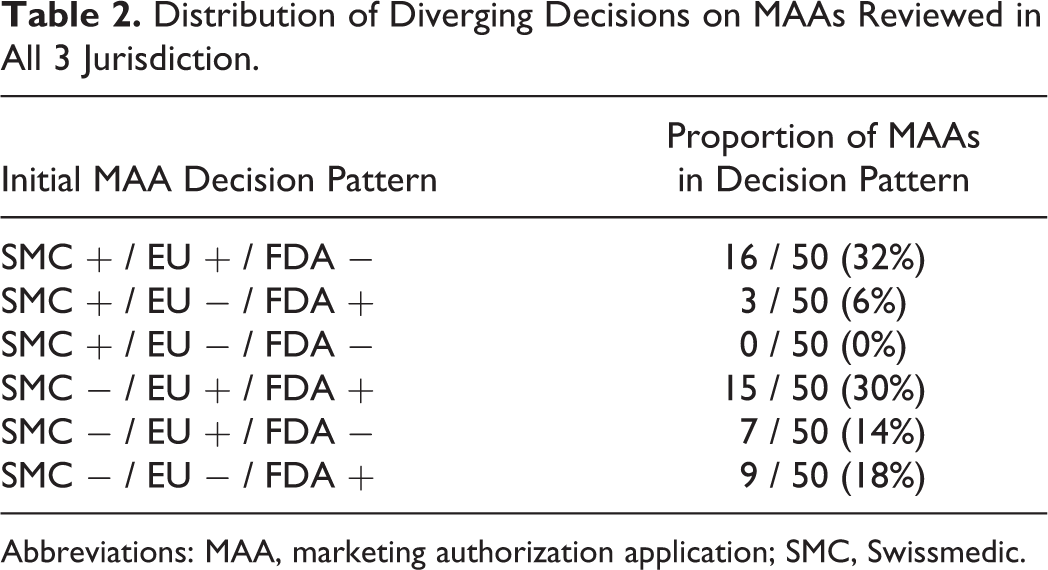
Abbreviations: MAA, marketing authorization application; SMC, Swissmedic.
Decision consensus between Swissmedic and the EU
Regarding the 255 MAAs independently reviewed in all 3 jurisdictions, decision consensus between SMC and EU was 90%: 211 (83%) of the MAAs were independently approved and 19 (7%) independently rejected by both respective authorities. In 3 cases (1%), SMC had approved a product that had been reviewed but was never authorized in the EU. Overall, 22 products (9%) were authorized by the EU but not by SMC (Figure 2).
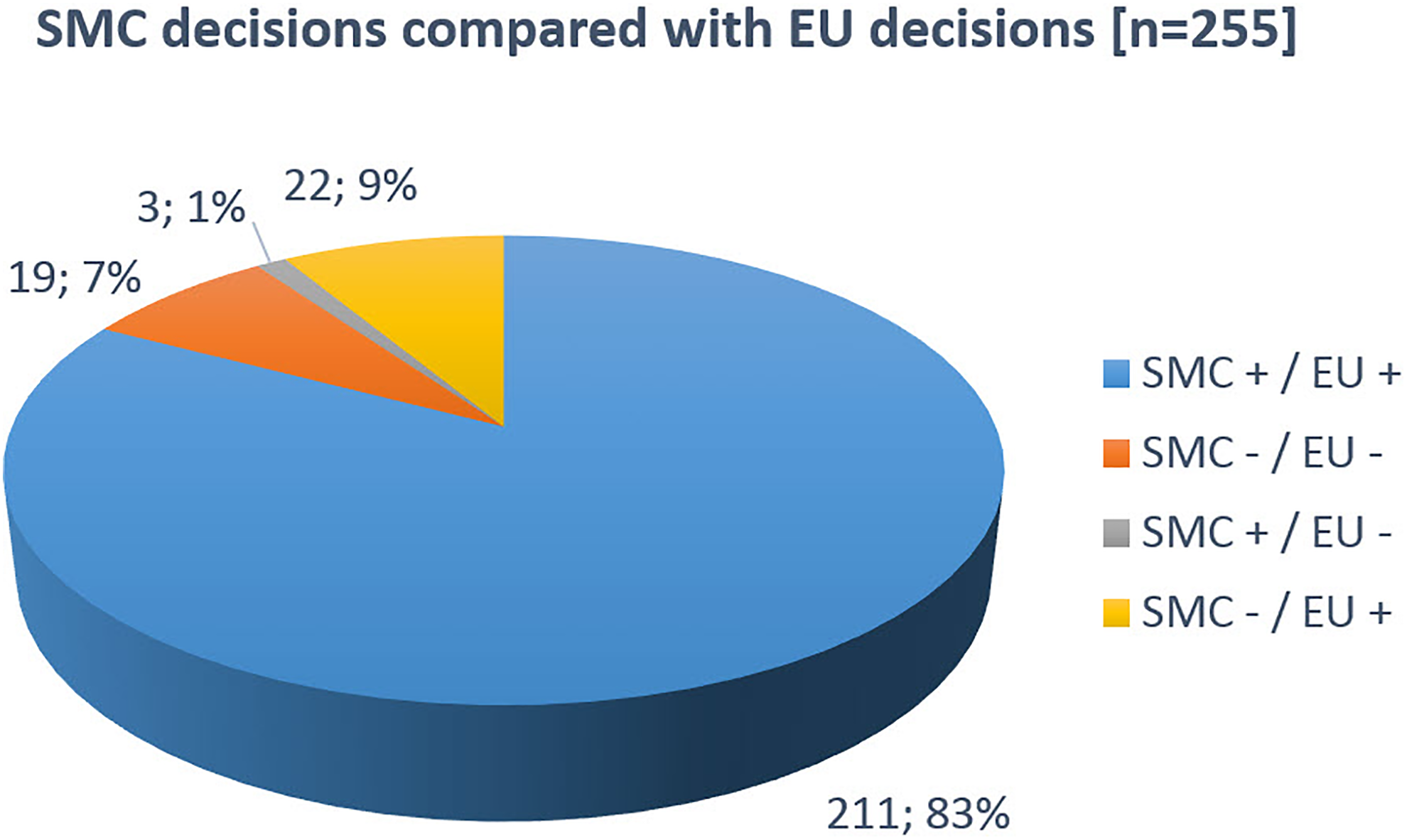
SMC decisions in comparison with EU decisions. SMC, Swissmedic.
We obtained similar results when the analysis comprised all MAAs reviewed by SMC and the EU, independent of their review or decision status in the USA (data not shown). Of the 214 MAAs approved by SMC, 211 (99%) were also approved in the EU: 182 (85%) were authorized through the EMA CP, 26 (12%) through the EU DCP/MRP, and 3 (2%) through a national procedure in at least 1 EU country. Of the 3 MAAs with negative outcome in the EU, 2 were refused and 1 application was withdrawn by the applicant upon a negative opinion by EMA’s Committee for Medicinal Products for Human Use (CHMP). Of the 41 MAAs with negative outcome at SMC, 19 (46%) were assessed but never approved through EMA’s CP, 18 (44%) were approved through EMA’s CP, and 4 (10%) were approved through the DCP/MRP process.
Decision consensus between Swissmedic and the FDA
Decision consensus between SMC and FDA was 85%: 198 (78%) of the MAAs were independently approved, and 17 MAAs (7%) were independently rejected by both respective DRAs. In 16 cases (6%), SMC approved a product that was assessed but never authorized by the FDA. In 24 cases (10%), MAAs were granted by the FDA but not by SMC (Figure 3).
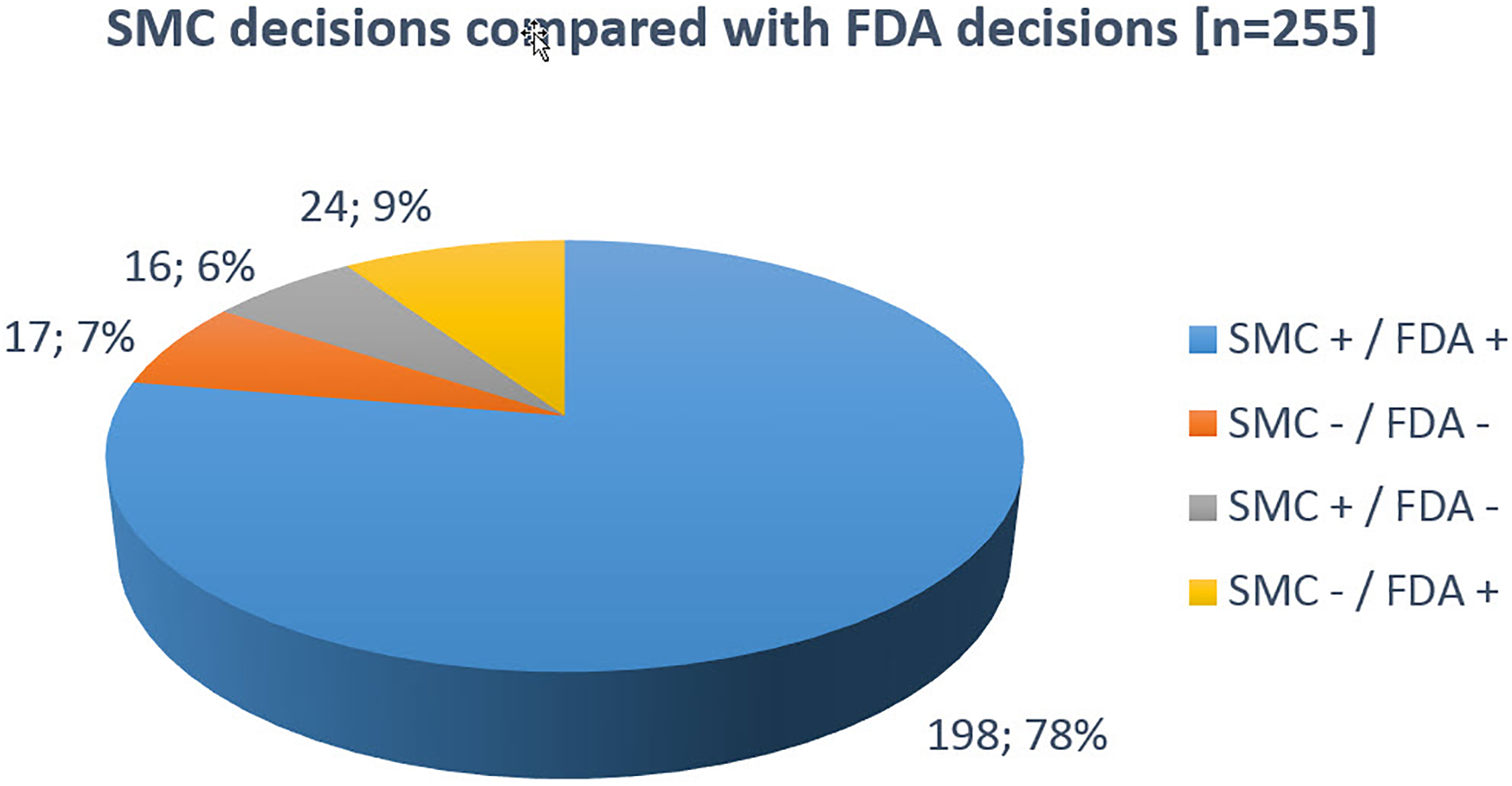
SMC decisions in comparison with FDA decisions. SMC, Swissmedic.
Of the 214 MAAs approved by SMC, 198 (93%) were also approved by the FDA and 16 (7%) MAAs were unilaterally rejected by the FDA. Of the 41 MAAs with negative outcome at SMC, 17 were also rejected by the FDA (41%). For 24 MAAs (59%), decisions diverged and MAs were granted in the USA only.
Decision consensus between the EU and the FDA
Decision consensus between EU and FDA was 86%: 210 (82%) of the MAAs were independently approved and 10 MAAs (4%) were independently rejected by both DRAs. In 23 cases (9%), the EU approved a product that was reviewed but never authorized by the FDA (Figure 4).
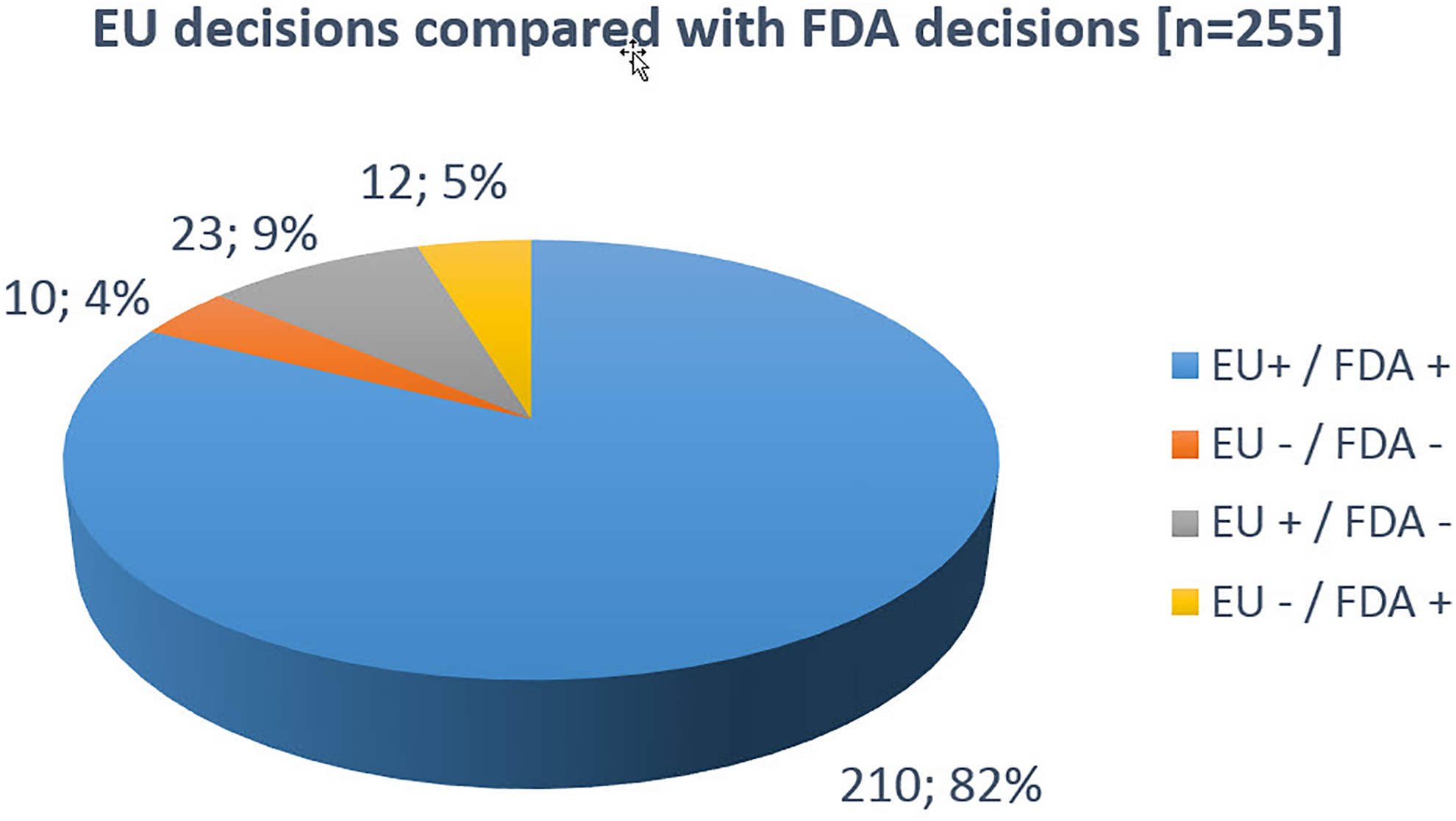
EU decisions in comparison with FDA decisions.
Of the 233 MAAs approved in the EU, 210 (90%) were also approved by the FDA; 23 (10%) MAAs were unilaterally rejected by the FDA. Regarding the 22 MAAs never authorized in the EU, in 10 cases (45%) rejections were also issued by the FDA. In 12 cases (55%), the FDA outcome diverged from the EU and the marketing authorizations were unilaterally granted by the FDA.
Postmarketing Withdrawals
When the benefit-risk balance of innovative medicinal products is judged differently by DRAs, an increased likelihood for risks and uncertainties may be associated with them. Real-world postmarketing data can tip the benefit-risk balance to the negative, and the marketing authorization holder must at times withdraw their medicinal product from the market. In this context, we investigated if the likelihood of postmarketing withdrawal correlates with MAA decision patterns of the 3 jurisdictions.
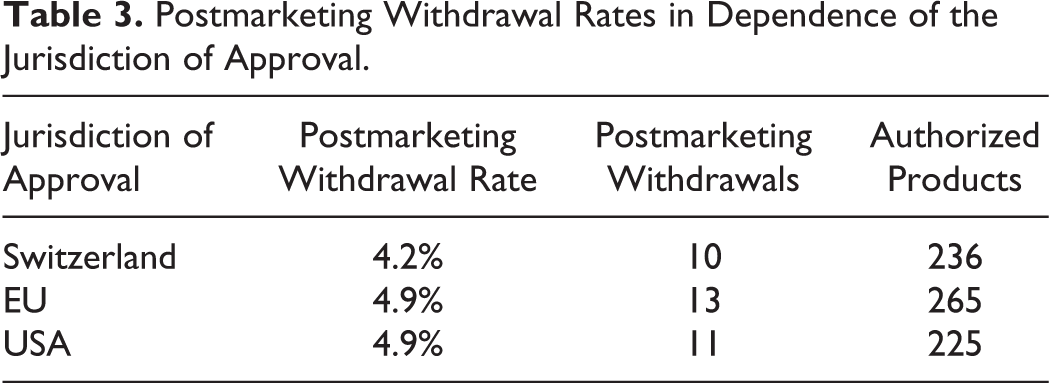
Two hundred eighty of the 298 MAAs that met the inclusions criteria were authorized in at least one of the 3 jurisdictions. Of these 280 authorized products, 16 were withdrawn postmarketing in at least one of the jurisdictions yielding an overall withdrawal rate of 6%. In Switzerland, 10 of the 236 (4.2%) MAAs originally approved by SMC were withdrawn postmarketing. In 3 cases, the reason for Swiss market withdrawal was safety concerns and in 8 cases the reasons were according to the marketing authorization holder of commercial nature or unknown to SMC. The 2 medicinal products withdrawn from the Swiss market for safety reasons had also been approved in the EU and were simultaneously withdrawn from the EU market. In contrast, the FDA had reviewed but never authorized these 2 products. The number of withdrawals in the EU and the USA was similar to those of Switzerland (Table 3). The reasons for postmarketing withdrawal after marketing are provided in Table 4.
Postmarketing Withdrawal Rates in Dependence of the Jurisdiction of Approval.
Reasons for Withdrawal.
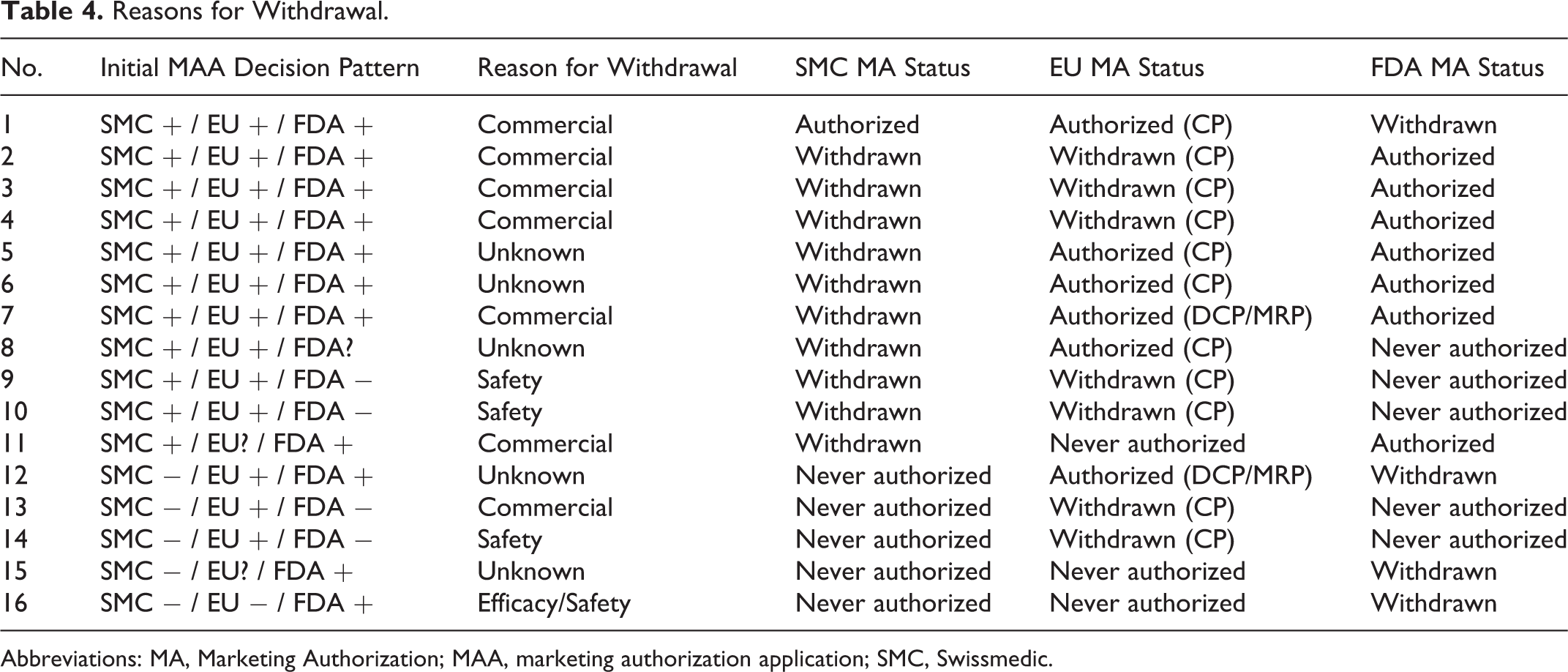
Abbreviations: MA, Marketing Authorization; MAA, marketing authorization application; SMC, Swissmedic.
The withdrawal rates for MAAs unanimously approved in all 3 jurisdictions was clearly lower (3.6%) than for MAAs with diverging outcome (11.1%). Notably, withdrawal rates varied in dependence of the jurisdiction where the MAAs were rejected in the first place: Of the 44 MAAs rejected by SMC but approved in the EU and/or the USA, 5 (11.4%) were later withdrawn from the market. In contrast, of the 23 MAAs with negative outcome in the USA but approved in Switzerland and/or the EU, 4 products (17.4%) were later withdrawn from the market. Of the 16 MAAs standalone approved in only one of the 3 jurisdictions, 3 had to be withdrawn postmarketing, yielding an even higher withdrawal rate of 18.8% (Table 5).
Withdrawal Rates in Dependence of DRA Decision Pattern.
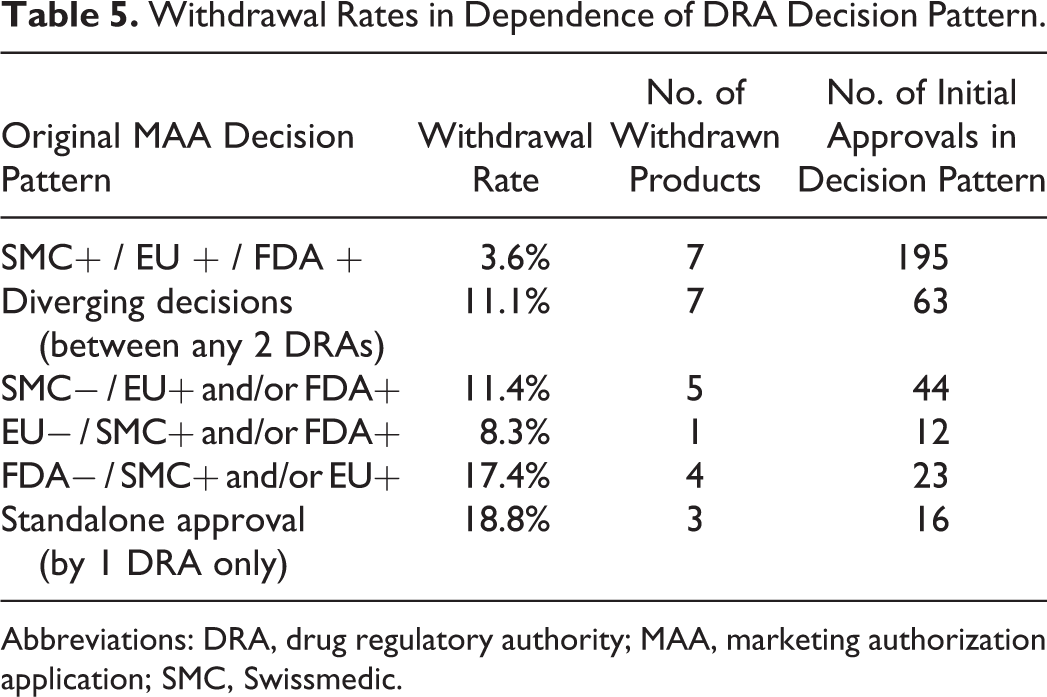
Abbreviations: DRA, drug regulatory authority; MAA, marketing authorization application; SMC, Swissmedic.
Swissmedic Reasons for Rejection
We investigated the reasons for rejection of the 40 MAAs with negative outcome in Switzerland but approval in either the EU or the USA. We pursued to identify concerns leading to rejection and to reveal if there was any indication that particular aspects in SMC’s approach to assessment differ from those of the EU and FDA authorities.
In a first step, we investigated the 40 SMC preliminary or official rejection letters regarding MAAs approved by either the EU (CP and DCP/MRP only) or the FDA. We recorded 114 therein stated concerns substantiating the SMC reasons for rejections—an average of 2.8 concerns per MAA.
For 28 of the 40 analyzed SMC rejections (70.0%), at least 1 clinical efficacy concern was raised, and for 25 of the same 40 rejections (62.5%), at least 1 clinical safety concern was raised. The predominant concerns communicated as a reason for rejection were “Marginal / no clinically relevant efficacy” (55%) and “Serious adverse events,” respectively (45%) (Table 6).
Swissmedic Reasons for Rejection Regarding Products Approved by the EU or FDA.a
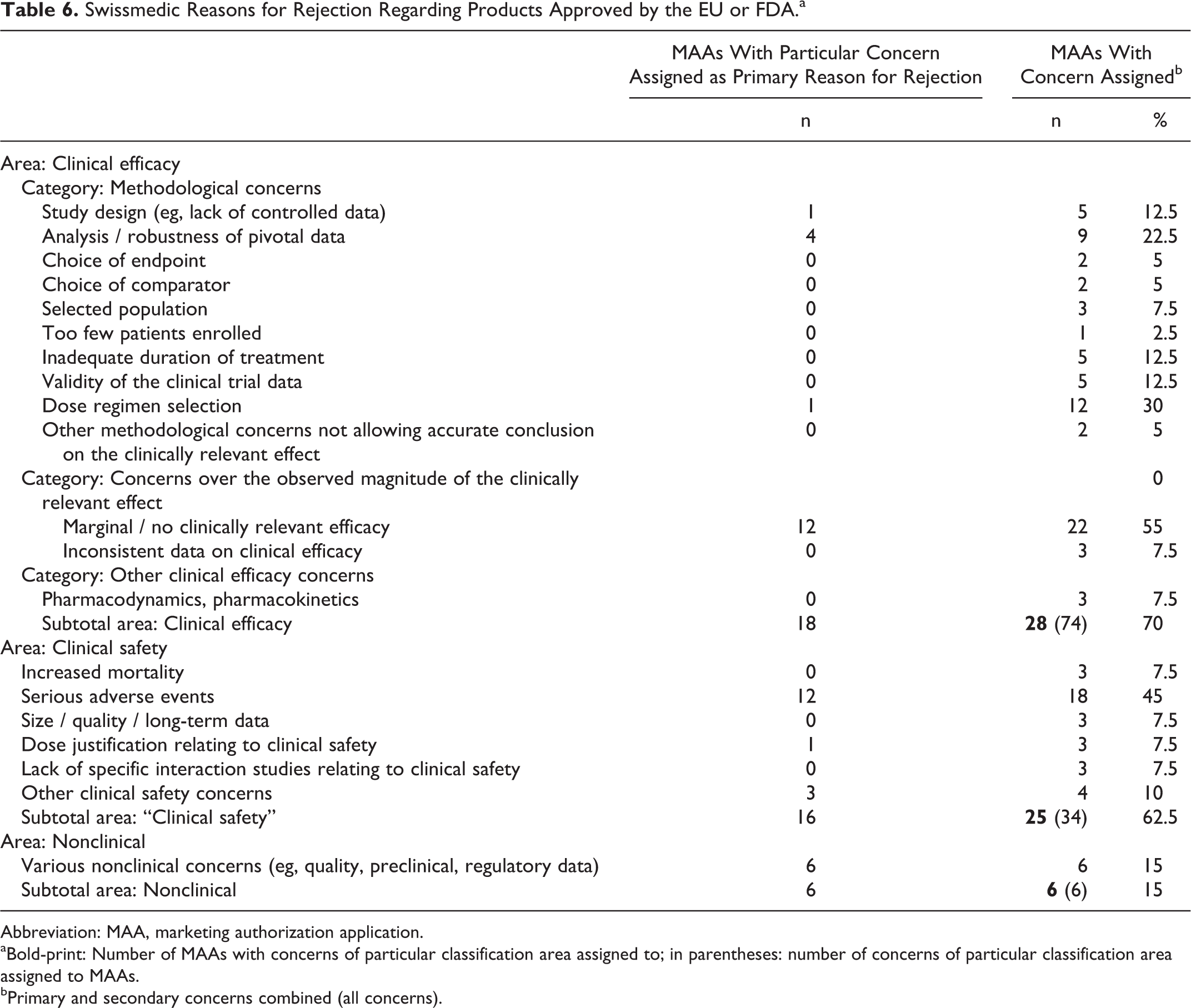
Abbreviation: MAA, marketing authorization application.
aBold-print: Number of MAAs with concerns of particular classification area assigned to; in parentheses: number of concerns of particular classification area assigned to MAAs.
bPrimary and secondary concerns combined (all concerns).
We also attempted to identify a “primary,” predominant reason for rejection for each of the 40 MAAs with negative outcome at SMC but authorized in the EU and/or USA.
For 18 (45%) of the 40 studied MAAs, the primary SMC concern giving grounds for rejection was “Clinical Efficacy,” for 16 (40%) MAAs, it was “Clinical Safety,” and for 6 (15%) MAAs it was various other, nonclinical concerns related to quality or preclinical data or other regulatory issues (Figure 5).
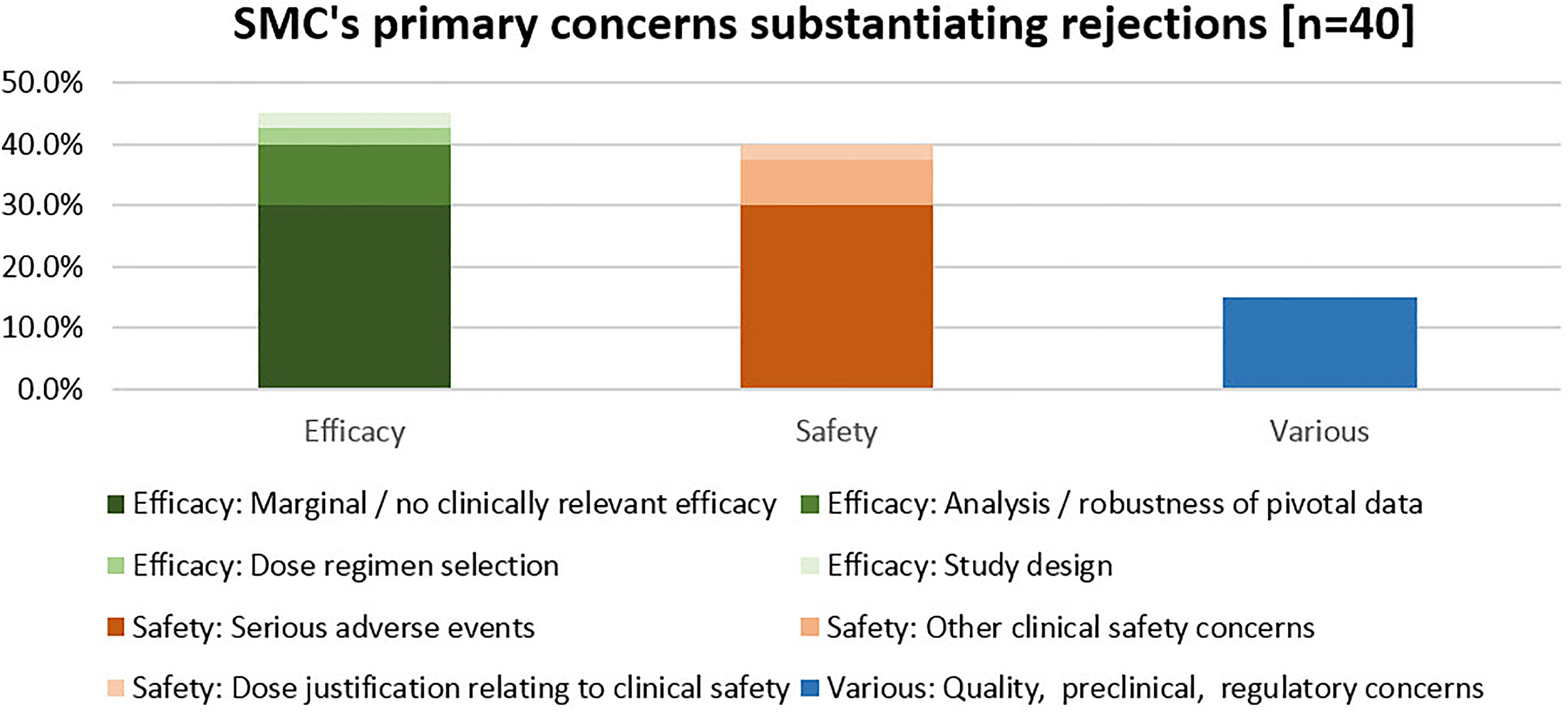
SMC’s primary concerns substantiating rejections. SMC, Swissmedic.
The primary clinical efficacy concerns divided into 12 cases of “failed drugs” where the data indicated insufficient efficacy and 6 cases of “failed development programs,” where an accurate conclusion regarding the clinical efficacy of the requested indication was not possible. Regarding clinical safety, the most frequent concern was “Serious adverse events,” accounting for 12 of the 16 (75%) primary safety concerns.
In 6 (15%) of 40 cases, the primary grounds for rejection were not clinical: in 1 case the company did not fulfill SMC’s labeling requirements leading to withdrawal of the application; in 3 cases the applications were withdrawn before the SMC List of Questions milestone was reached (once because of newly disclosed serious quality issues, twice because the respective MAAs were withdrawn in the EU after major concerns had prevented a marketing authorization). One MAA could have been authorized from a clinical and preclinical point of view but serious quality issues remained unresolved, in 1 case serious concerns over preclinical toxicology lead to a negative preliminary decision and subsequent withdrawal of the application, and in 1 last case other regulatory reasons were decisive.
For the 3 cases of positive outcome at SMC but negative outcome in the EU, the CHMP’s primary concerns relating to rejection were in 2 instances clinical safety concerns (“Other serious adverse”) events. In the third case, the CHMP’s primary concern was the inadequate study design: the clinical data did not allow the CHMP to properly assess the benefit of the medicine, because comparison with any other treatment or placebo in another group of patients was lacking (“Study design, e.g., lack of controlled data”). SMC shared these concerns, leading to a negative opinion in the EU but in contrast SMC concluded that the benefits still outweighed the risks and that therefore a marketing authorization was granted in Switzerland. FDA’s 16 negative decisions diverging from SMC positive decisions could not be investigated because in case of refusal or applicant withdrawal, the FDA’s data are not in the public domain.
Discussion
Of the 298 MAAs assessed by SMC, the great majority were also reviewed in the EU (98%) and the FDA (87%). We did not examine the extent to which MAAs filed at the FDA or in the EU were also submitted to SMC, but analysis of public records supports the notion that innovative new products are also brought to market in Switzerland. Although the Swiss market size is much smaller than the EU’s or the USA’s, Swiss marketing authorization can serve as a reference for abridged procedures in a large number of developing and emerging countries. Hence the value of a Swiss marketing authorization goes well beyond the domestic market.
One of the primary aims of this study was the comparison of the DRA’s overall decision patterns. There was a high degree of convergence between the decisions of SMC and EU (90%) or FDA (85%). However, in the cohort of decisions diverging between SMC and the EU, MAAs were predominantly approved in the EU (88%). In contrast, when decisions diverged between SMC and the FDA, negative and positive outcomes were more equally distributed as compared to the SMC-EU comparison. The decision consensus rate of 86% between EU and FDA was placed in between the SMC-EU and SMC-FDA consensus rates. Interestingly, similar to the SMC-EU comparison, in the cohort of decisions diverging between EU and FDA, it was in the large majority in the EU where MAAs were approved (Table 7).
Comparison of SMC, EU, and FDA Marketing Authorization Decisions on Applications Reviewed in All 3 Jurisdictions.

Abbreviations: DRA, drug regulatory authority; SMC, Swissmedic.
Remarkably, 20% of the considered MAA decisions diverged between the DRAs. With an approval rate of 84%, SMC appears to be slightly more stringent than the EU and the FDA, which approved 91% and 87% of the MAAs, respectively. For an adequate interpretation of these findings, a number of confounding factors should be considered.
First, regulatory decisions depend to some extent on the timing of MMA filing. SMC tends to receive MAA later than the EMA or the FDA 12 and as consequence the body of evidence regarding safety and efficacy submitted to SMC might be slightly more comprehensive compared to earlier filings at the EMA or FDA. Although the timing might therefore play a role, a preliminary analysis of our data at least gave no indication that SMC regulatory decisions were biased by EMA or FDA decisions known to SMC at the time point that decisions were made (data not shown).
Second, medicinal products containing the same NME may be authorized for slightly different indications across jurisdictions, making a direct comparison challenging. This issue was recently addressed by Dörr et al, showing that the majority of initial labeling differences resulted from writing styles or structures of the Summary of Product Characteristics (SmPC) and were actually not clinically relevant. 12 Accordingly, the effect on MAA decisions should be minor.
Third, the application of FDA’s special designation programs such as “Priority Review” and “Accelerated Approval” is associated with an increased likelihood of medicinal products being approved in the first review cycle. 13 The more closely a DRA is involved in the strategy and design of the clinical development program, the higher appears to be the probability of the medicinal product eventually being approved. Thus, in the future, expedited programs and special designations such as EMA’s PRIME scheme and FDA’s Breakthrough Therapy and Fast Track designations could further improve regulatory success rates. For this study, however, we did not consider parameters regarding the applied regulatory pathways and we could therefore only speculate about the magnitude of such an effect.
Last but not least, cultural, political, legal, as well as country-specific health priority aspects may in individual cases have tipped the balance toward approval or rejection. The judgment of the benefits and risks of a medicinal product is not an exact science but will to some extent also depend on the personalities and backgrounds of the colleagues involved in the assessment processes. In order to ensure a well-balanced decision-making process, the DRAs we investigated have established peer review procedures and, additionally, external scientific expert committees. Despite these measures, a subjective element in the evaluation process cannot entirely be excluded. For instance, potential bias can be introduced by articulate “internal opinion leaders” with a certain standing within their respective organizations or by subconscious groupthink mechanisms.
Analysis of the withdrawal rate provided remarkable results regarding the postmarketing performance of approved products. The lack of decision consensus between the chosen DRAs is associated with a 3-fold increased likelihood of a particular medicinal product to be withdrawn postmarketing (Table 5). Moreover, postmarketing withdrawal rates differed depending on the jurisdiction where the particular products had been rejected in the first place: Particularly, if the FDA had initially rejected a product for which marketing authorization was granted in the EU or in Switzerland, there was a 17.4% probability for the product to be withdrawn from the market at a later point. Although the numerical values of this analysis were low, the results are still striking considering the potential health implications associated with withdrawal of a medicinal product.
Our analysis of the reasons for rejection demonstrate that the 2 areas of SMC concern communicated to the applicant as the primary, decisive reasons for rejection were on balance efficacy (45%) and safety (40%) (Figure 5), that is, the main issues relating to the benefit/risk evaluation. The results provided no evidence that SMC’s scientific assessment is biased in its weighting of safety and efficacy. Interestingly, for a relatively high number of rejected MAAs (55%), SMC had also expressed concern about methodological shortcomings related to testing of clinical efficacy, and in 6 cases (15%) the lack of substantial evidence to conclusively judge the efficacy of the medicinal product was even the primary (decisive) reason for SMC’s rejection (Table 6). Thus, although demonstration of efficacy and safety are naturally the major determinants for a successful MAA, methodological concerns still played a surprisingly significant role.
Conclusion
With a high overall decision consensus of 84% to 90%, the DRAs generally came to the same overall conclusions regarding the benefit-risk balance of the assessed medicinal products. This finding is supported by the notion that the 3 DRAs’ approach to reviewing MAAs should be very similar: SMC, the EMA, and the US FDA are all ICH member states and therefore adhere to the same set of scientific guidelines and pharmacopeial standards regarding all critical aspects of development.
When divergence in regulatory decisions occurred, they were about equally distributed among the DRAs. Remarkably, medicinal products with a history of diverging decisions also had a measurably increased probability of postmarketing withdrawal for being ineffective or unsafe. However, in order to obtain a more refined picture of the postmarketing performance of medicinal products, more common risk-mitigating regulatory measures such as restrictions of therapeutic indication or incorporation of additional safety warnings to the SmPC should be addressed.
So how to explain the 20% diverging decisions? Taking into account slightly different time windows for submission, the possible impact of special designation programs 13 and possibly a residual subjective element, the remaining differences may be explained by cultural and legal context, as well as public health priorities at the time of assessment. These aspects should be subject of future research into diverging decisions of major DRAs and how they are governed in their respective jurisdictions.
Footnotes
Acknowledgments / Disclaimers
The views and opinions expressed in this article are those of the authors and should not be attributed to the Swiss Agency for Therapeutic Products.
Conferences / Posters
Poster presentation at 28th Annual EuroMeeting 2016, DIA, Hamburg Germany
Declaration of Conflicting Interests
No potential conflicts were declared.
Funding
No financial support of the research, authorship, and/or publication of this article was declared.