
Abstract
The GNE gene encodes the UDP-GlcNAc-2-epimerase/ManNAc kinase, a bifunctional enzyme required for the synthesis of sialic acid. The mouse Gne gene is essential for embryonic development, but humans with recessive partial loss of function GNE mutations can develop infantile thrombocytopenia, juvenile amyotrophic lateral sclerosis, or adult-onset myopathy (GNE myopathy). We have created inducible Gnelox/lox gene deletion mice to study how loss of Gne in adult mice relates to these disease states. Systemic Gne gene deletion in tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice caused uniform fatality within 30 days of gene deletion with spontaneous bleeding, thrombocytopenia, and anemia. Skeletal myofiber-specific Gne deletion in tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice had no bleeding and no muscle pathology at 60 or 270 days post-treatment. Intramuscular injection of AAV.MCK.GFP-Cre in Gnelox/lox mice also showed little to no evidence of muscle pathology, while AAV.CMV.GFP-Cre caused extensive muscle damage, reduced muscle force, and changed expression of markers for muscle regeneration, muscle cell senescence, muscle denervation, and muscle atrophy. These data demonstrate that Gne is an essential gene in adult mice that can mimic aspects of human hematologic and muscle diseases caused by GNE mutations, but suggests induction of muscle disease requires loss of gene GNE expression in cell types beyond skeletal myofibers.
Introduction
The GNE gene encodes the UDP-GlcNAc-2-epimerase/ManNAc kinase, a dual function enzyme responsible for the committed steps in sialic acid (Sia) biosynthesis.1–4 Deletion of mouse Gne in zygotes results in embryonic lethality, 5 and no recessive null human GNE mutations have been described. Humans presenting with homozygous or compound heterozygous recessive GNE mutations, most often missense mutations, can develop a variety of seemingly tissue-specific diseases that present at different ages. Infants can present with a severe bleeding disorder involving thrombocytopenia with reduced platelet count.6–12 Teenagers can present with juvenile amyotrophic lateral sclerosis (ALS), 13 a motor neuron disease, or with peripheral motor neuropathy.14,15 More commonly, adults can present with GNE myopathy (GNEM), a severe muscle disease characterized by progressive muscle weakness and muscle atrophy.16–18
The type of disease presentation, and the age of onset, correlate in part with the specific GNE mutations found in patients. 19 Over 260 mutations in GNE are known to cause disease, and some mutations are more prevalent in certain ethnic groups; for example, M743T mutations show high GNEM incidence in Iranian Jews, D207V and V603L mutations in Japanese, I618T in Romas, and V727M in Indians.16,17,20–25 H186R, D239Q, and H705R mutations have been reported in patients with ALS and motor neuropathy,13–15 while C594Y, P735R, T417M, R420Q, D207V, V603L and G739S mutations have been associated with thrombocytopenia and sleep apnea.7,8,26–28 While GNEM was originally thought to be a rare genetic disease, a recent study of known GNEM mutations in large genetic databases estimated a disease incidence between 11.0 and 87.6 cases per million, about 10 times higher than originally thought. 29
The complexity of human disease genetics for GNE begs the question of whether these seemingly disparate disease presentations share a common cellular mechanism, as the GNE gene, and sialic acid, are expressed in all tissues of the body. Indeed, many GNEM patients have thrombocytopenia, albeit often with levels of platelet reduction that do not cause spontaneous bleeding.26,28,30 Similarly, muscle atrophy in GNEM may be related to motor neuron disease as is found in GNE-dependent ALS or motor neuropathy, as denervation of motor neurons from muscles results in muscle atrophy. 31 In addition, Gne haploinsufficiency has impacts on muscle and brain aging; Gne+/− mice have reduced ambulation, increased synapse loss, and increased neurodegeneration as they age.32,33 These findings demonstrate that loss of GNE function may lead to complex disease phenotypes that may differ depending on the extent of reduced GNE gene function in particular tissues. As Gne is essential for mouse embryogenesis, 5 we have made inducible Gne deletion mice (Gnelox/lox) to study loss of Gne function in adult mice.
Results
Gne is an essential gene in adult mice
CRISPR-Cas9 methods were used to delete exon 3 of the mouse Gne gene (GenBank:NM_001190414). A description of all mouse lines used in this study is given in Table S1. Gne+/− mice were interbred, and no Gne−/− mice were born from 16 litters comprising 122 total offspring (Table S2). 30 or 31 offspring would have been expected if Gne−/− mice were to have normal viability. This result replicates previous embryonic stem cell gene deletion experiments demonstrating lack of viability with embryonic Gne gene deletion. 5
We created Gnelox/lox mice by inserting two loxP sites that flank exon 3 using CRISPR-Cas9 methods in zygotes, where introduction of the Cre recombinase could cause exon 3 deletion (Figure S1). This deletion would be for exon 3 in both the mGne1 and mGne2 splice forms variants of the mouse gene. 34 The mGne1 transcript begins in exon 2 and expresses exon 3, while mGne2 transcript begins in exon 1, has exon 2 spiced out, and expresses exon 3. 34 The deletion of mouse exon 3 would also be equivalent to exon 3 deletion in the hGNE1 and hGNE2 transcript variants of the human gene. 35 Gnelox/lox mice were bred to transgenic mice expressing an inducible Cre recombinase-estrogen receptor (Cre-ERT2) fusion protein driven by the Reverse Orientation Splice Acceptor 26 (Rosa) gene trap locus, 36 which yields broad tissue transgene expression, or by the human alpha skeletal actin promoter (HSA), 37 which yields skeletal myofiber-specific transgene expression. Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice were made bearing the transgene locus on both alleles to increase CreERT2 transgene expression, while HSA-CreERT2/+Gnelox/lox were made bearing a single copy of the transgene locus. Expression of the CreERT2 fusion protein allows Cre to be sequestered in the cytoplasm due to its fusion to ERT2, where it will have no gene deletion activity. When a hormone such as tamoxifen (Tam) is added, binding of tamoxifen to the ERT2 protein induces migration to the nucleus, where Cre recombinase can then induce deletion of loxP-flanked DNA regions.38,39
Tamoxifen was given to adult, 3–4-month-old, Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice, once daily for five days at a dose of 75 mg/kg. Exon 3 of the Gne gene was deleted to some degree in all tissues tested, including spleen, lung, kidney, colon, thymus, liver, brain, heart, and skeletal muscles (Fig. S2B). No exon 3 Gne deletion was observed in untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox or Gnelox/lox mice (Figures S2A,S2E). Tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice showed exon 3 Gne deletion only in skeletal muscles (Figure S2D), while untreated HSA-CreERT2/+Gnelox/lox mice showed no deletion (Figure S2C).
Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice treated with tamoxifen showed uniform mortality by 14 days post-tamoxifen treatment in male mice and by 30 days in female mice (Figure 1). Half of male mice perished by 8 days post-treatment. Tamoxifen treatment of mixed gender HSA-CreERT2/+Gnelox/lox mice showed no lethality for up to 240 days (Figure 1) and there was no lethality in the first 30 days for tamoxifen treatment of Rosa-CreERT2/Rosa-CreERT2 mice (ns). Statistical significance between groups, determined by log-rank Mantel-Cox test, showed a difference in survival between male and female tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice (p < 0.01) and a significant difference between both of these groups and tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice (p < 0.0001 for both comparisons). There was no death in untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox, HSA-CreERT2/+Gnelox/lox, and Gnelox/lox mice for up to 9 months of age (ns).
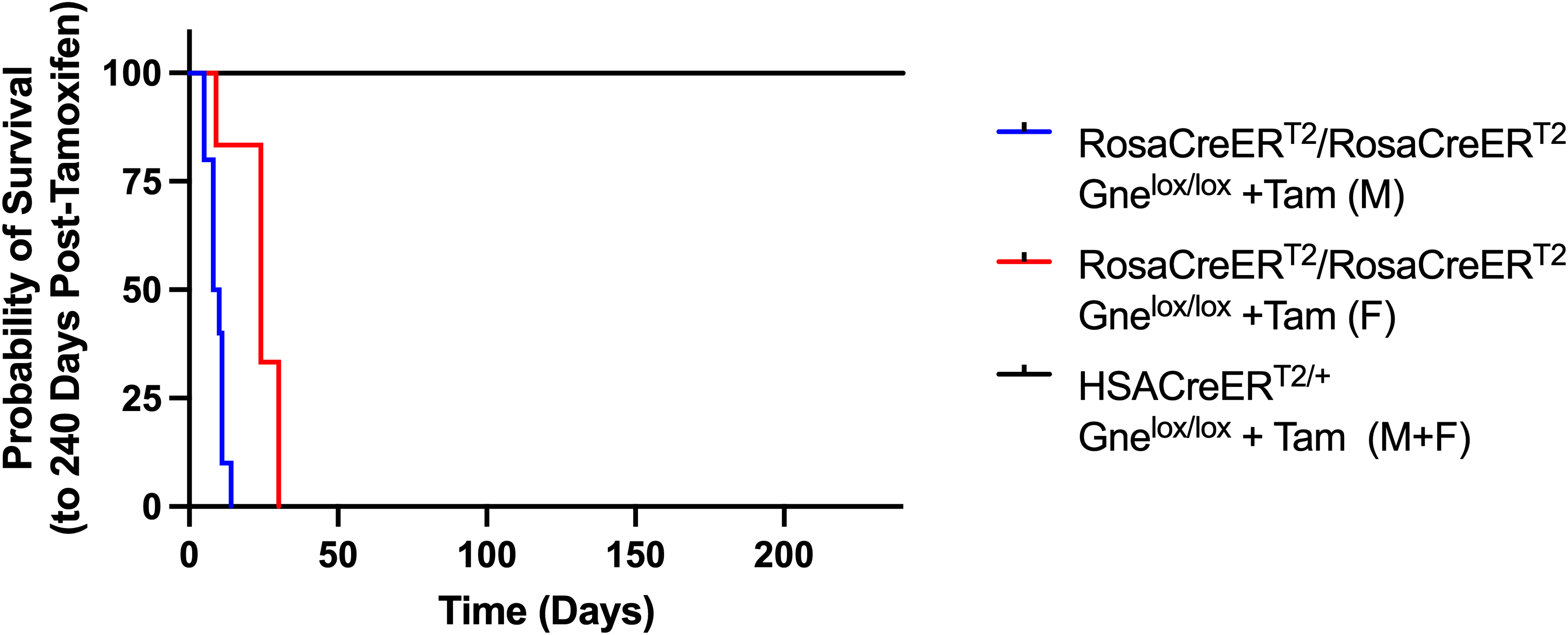
Kaplan-Meyer survival curve after systemic or muscle-specific Gne gene deletion in adult mice. Tamoxifen (+Tam) was given to Rosa-CreERT2/Rosa-CreERT2Gnelox/lox or HSA-CreERT2/+Gnelox/lox mice to induce Gne gene deletion systemically or in skeletal myofibers, respectively. Mice were given tamoxifen at between 3 and 4 months of age. Time is counted from the first day after the five-day tamoxifen regimen was given. Survival of untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox male (blue, n = 10), Rosa-CreERT2/Rosa-CreERT2Gnelox/lox female (red, n = 6), and mixed gender HSA-CreERT2/+Gnelox/lox mice (black, n = 11; 3 females and 8 males) was compared for up to 240 days total.
Because of the speed with which mortality occurred, reductions in Gne gene expression varied amongst tissues. At endpoint, Gne gene expression in tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice was dramatically reduced, to below 1% of normal in liver, spleen, and colon and to or below 10% of normal in kidney, lung and thymus (Figure S3A). Gene expression changes in muscle and brain, however, were smaller. Gne expression was reduced to 62% of normal in brain and to 35% of normal in heart (Figures S3A, S3B). In skeletal muscles, Gne gene expression was reduced to 21% of normal in diaphragm but only to 71–80% of normal in tibialis anterior (TA), gastrocnemius (gastroc), and quadriceps femoris (quad) (Figure S3B). For tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice (Figure S3C-F), Gne expression was only reduced in skeletal muscles - to 22–36% of normal in TA, gastroc, quad, and triceps and to 71% of normal in diaphragm at 60 days post-treatment (Figure S3D). At 270 days post-treatment, Gne expression was reduced to 21–29% of normal in limb muscles and to 76% of normal in diaphragm (Figure S3F).
Western blots of mouse Gne protein showed significant reductions in protein expression in liver and kidney in tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice (Figure S4). Because mouse Gne gene expression is very low in skeletal muscle relative to other organs, 40 we could not identify muscle protein expression by Western blotting, but measures of total sialic acid content in muscle from tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice was reduced by 40–50% compared to untreated controls, with a reduction in the percentage of bound Sia in the remaining pool as well (Figure S5A).
Systemic Gne deletion in adult mice induces anemia, thrombocytopenia, and spontaneous bleeding
Whole blood was analyzed near endpoint for counts of red blood cells (RBCs), hemoglobin, hematocrit, mean corpuscular hemoglobin, mean corpuscular volume, white blood cells (WBCs), and platelets (Figures 2 and 3). There was a minor effect of tamoxifen on Gnelox/lox mice and Rosa-CreERT2/Rosa-CreERT2 mice, neither of which had any exon 3 Gne deletion. Tamoxifen treatment of Gnelox/lox mice and Rosa-CreERT2/Rosa-CreERT2 mice led to a slight, but significant, reduction in RBCs, hemoglobin, and hematocrit, and to a larger drop in WBCs. These results in fully mature adult mice contrast with reports of tamoxifen effects in embryonic or neonatal Rosa-CreERT2/Rosa-CreERT2 mice, where severe anemia can be present even in the absence of target gene deletion.41,42 There was a further significant decrease in RBCs (Figure 2A), hemoglobin (Figure 2B) and hematocrit (Figure 2C) in tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice relative to untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice or to tamoxifen-treated Gnelox/lox or Rosa-CreERT2/Rosa-CreERT2 mice (Figure 2). This was true for both male and female mice at both time-points tested (1 or 5 days post-tamoxifen treatment in males and 7 or 28 days post-tamoxifen treatment in females). The degree of decrease was greater for tamoxifen-treated male mice at 5 days than at 1 day post-treatment, while values were equally low at 7 and 28 days for females. Both male and female Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice treated with tamoxifen had a 50% or greater reduction in RBCs, hemoglobin, hematocrit, and WBCs compared to untreated strain-matched controls. Changes in mean corpuscular hemoglobin (Figure 2D) and mean corpuscular volume (Figure 2E), by contrast, were most often statistically insignificant.
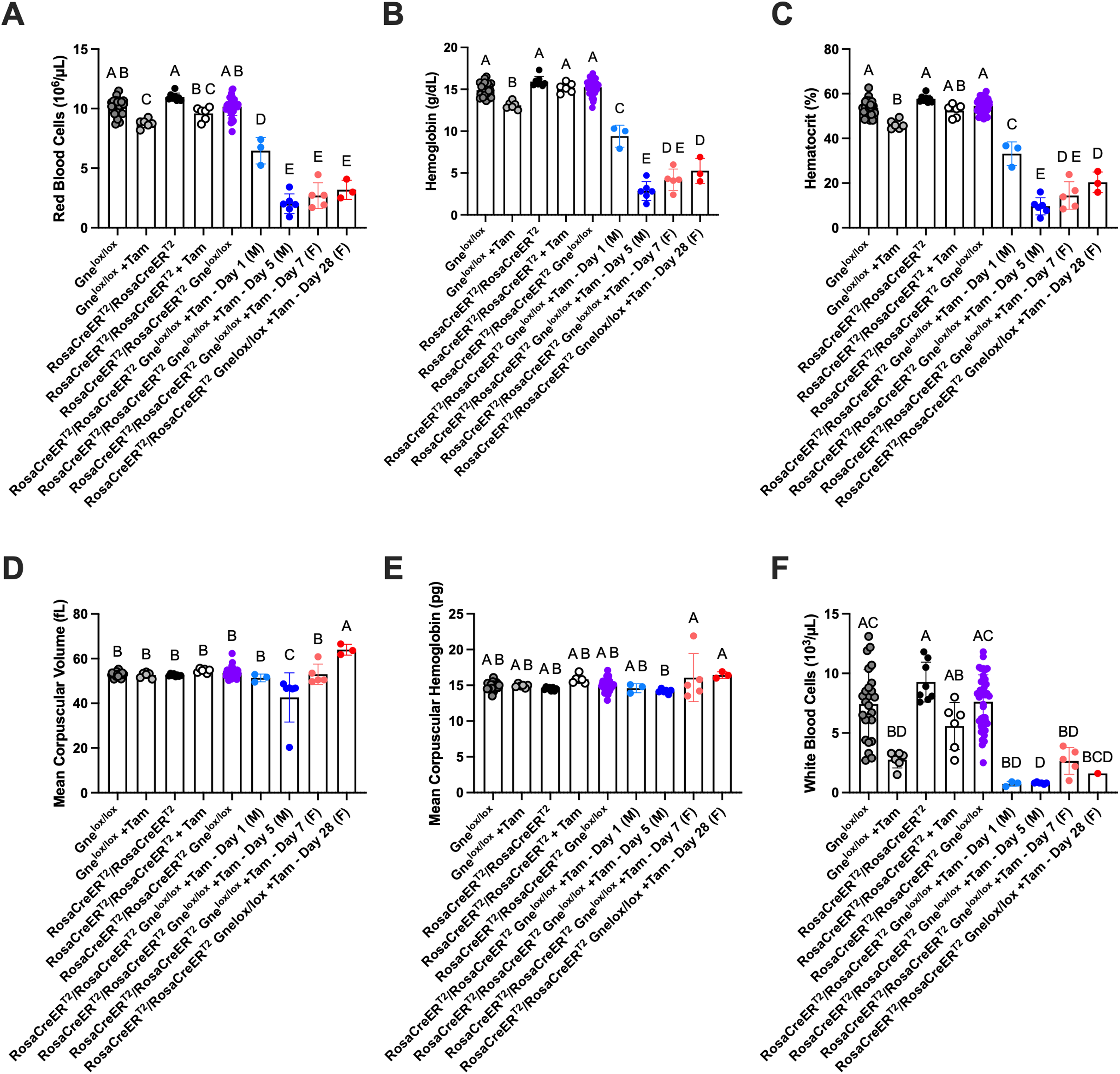
Anemia after systemic Gne gene deletion in adult mice. Whole blood counts at endpoint for red blood cells (A), hemoglobin (B), hematocrit (C), mean corpuscular volume (D), mean corpuscular hemoglobin (E), and white blood cells (F) are shown. Untreated and tamoxifen (+Tam)-treated Gnelox/lox, Rosa-CreERT2/Rosa-CreERT2, and Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice are compared. Rosa-CreERT2/Rosa-CreERT2 and Gnelox/lox mice shown are combined at 1-2 weeks for males and 3-4 weeks for females, while Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice are shown at 1 or 5 days for males (M) or 7 or 28 days for females (F) post-treatment. Errors are SD. Statistical significance between groups determined by one-way ANOVA with Tukey's post-hoc test showing differences of p < 0.05 between groups. Untreated Gnelox/lox n = 25; Gnelox/lox +Tam n = 6; untreated Rosa-CreERT2/Rosa-CreERT2 n = 8; Rosa-CreERT2/Rosa-CreERT2 +Tam n = 6; untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox n = 36–40; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox +Tam-Day1 (M) n = 3; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day5 (M) n = 6; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day7 (F) n = 5; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day28 (F) n = 3.
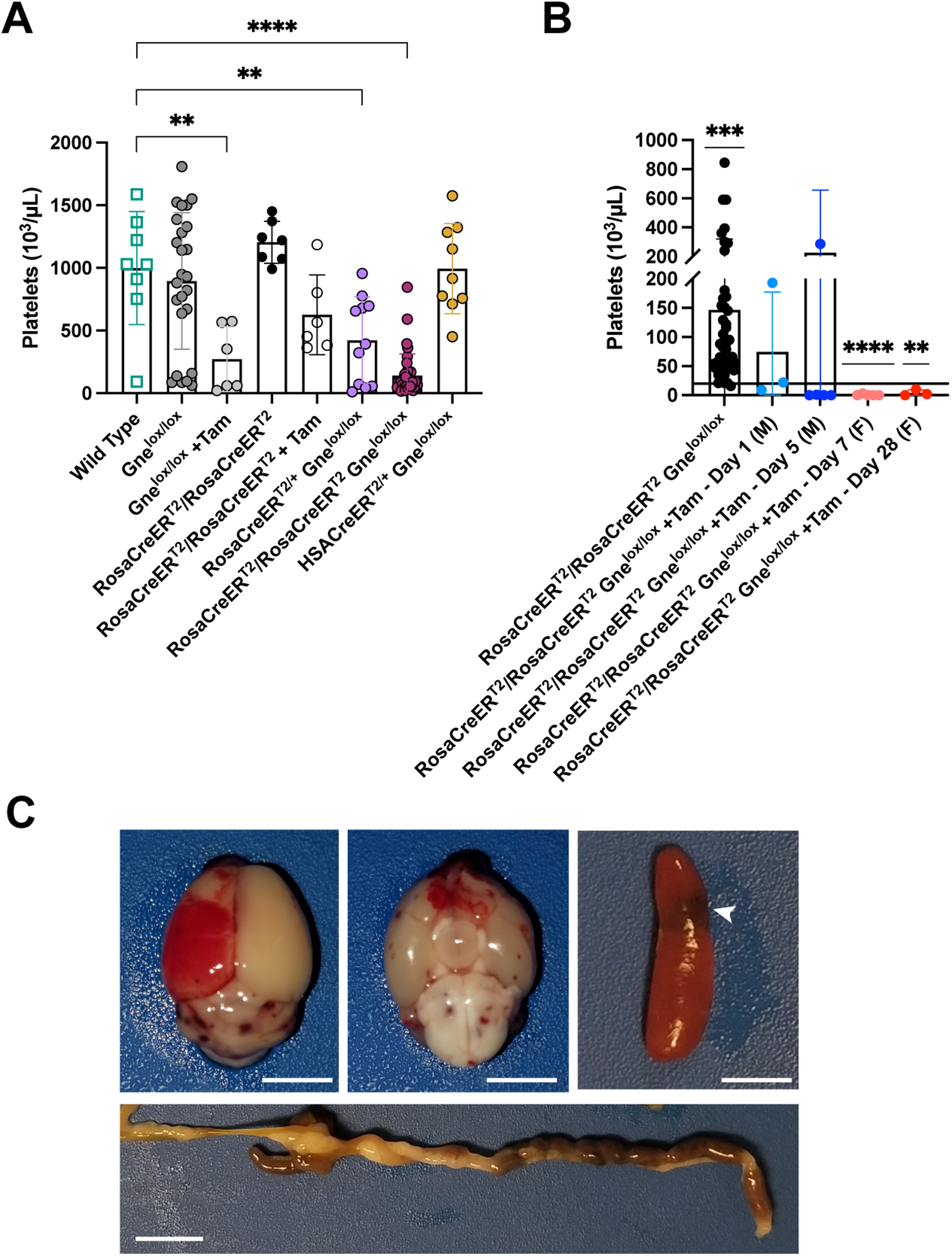
Thrombocytopenia and spontaneous bleeding after systemic Gne gene deletion in adult mice. (A,B) Whole blood cell counts of platelets are shown. (A) Platelet counts for control cell groups including mice with a single copy or with two copies of the Rosa-CreERT2 locus with Gnelox/lox (n = 10, n = 36, respectively), one copy of the HSA-CreERT2 locus with Gnelox/lox (n = 9), wild type mice (n = 8), and tamoxifen (Tam)-treated Gnelox/lox mice and Rosa-CreERT2/Rosa-CreERT2 mice (n = 7). (B) Comparison of blood platelet counts in untreated (n = 44) or tamoxifen (+Tam)-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice. Mice aged between 3 and 4 months were treated with tamoxifen (+Tam) and analyzed at 1 (n = 3) or 5 days (n = 6) (males, M) or 7 (n = 5) or 28 (n = 3) days (females, F) after the final treatment. Line shows a platelet value of 20 × 103/μL, a value below which spontaneous bleeding can occur. (C) Examples of spontaneous bleeding in tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox at endpoint in brain (top and bottom view, upper left and middle), spleen (at arrow, upper right), and intestine (below). Bars are 0.5 cm (brain, spleen) and 1.0 cm (intestine). Errors in A and B are SD. Statistical significance between groups determined by one-way ANOVA with Dunnett's post-hoc test vs. wild type (A) or by one-way sample t test versus a value of 20 with Wilcoxon test (B). Untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice are significantly above the value of 20, while 7 or 28 day treated female Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice are significantly below the value of 20. *p < 0.05, **p < 0.01, ****p < 0.0001.
There was a significant reduction in platelet count in untreated Rosa-CreERT2/+Gnelox/lox and Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice relative to wild type mice (Figure 3A). In addition, platelet levels were significantly reduced in tamoxifen-treated Gnelox/lox mice relative to wild type (Figure 3A). All but one of these values, however, did not fit the definition for severe thrombocytopenia (below 20 × 103/μL, www.merckmanuals.com/professional/multimedia/table/platelet-count-and-bleeding-risk) (Figure 3A), a level where spontaneous bleeding may occur without injury. There was no significant reduction in platelet count, relative to wild type, in untreated Gnelox/lox, Rosa-CreERT2/Rosa-CreERT2, or HSACreERT2/+Gnelox/lox mice (Figure 3A), suggesting that the platelet reductions in untreated Rosa-CreERT2/+Gnelox/lox and Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice were due to the presence of the combined Rosa-CreERT2 and Gnelox/lox loci. Almost all tamoxifen-treated male Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice at 1 or 5 days post treatment and all tamoxifen-treated female Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice at 7 or 28 days post-treatment had platelet levels below 20 × 103/μL (Figure 3B). By 1–2 weeks post-tamoxifen treatment in males or by 3–4 weeks post-treatment in females, the end of lifespan endpoint, there was clear evidence of spontaneous bleeding in multiple organs, including the brain, spleen, and intestine (Figure 3C). There was no obvious evidence of spontaneous bleeding in dissected kidneys or heart (ns), but hematoxylin and eosin staining of kidney sections suggested the presence of blood in some tubules (Figure S6), mimicking hematuria previously reported in GNEM743T/M743T knock-in mice. 43 Kidney sections showed increases in staining for lectins that only bind when terminal sialic acid is absent (PNA and ECA), particularly in glomeruli (Figure S7), while changes in MAA and SNA, which bind sialic acid directly, were not evident (Figure S8). Similar results were seen in cardiac sections as well (Figures S9, S10).
Three tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice showed an elevated pro-thrombin time (PTT) greater than 90 s (Figure 4A), and two tamoxifen-treated mice also showed elevated activated partial thromboplastin time (aPTT) greater than 139 s (Figure 4B). Fibrinogen levels were reduced somewhat by tamoxifen treatment in Gnelox/lox mice, and this was not significantly reduced further in treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice (Figure 4C).
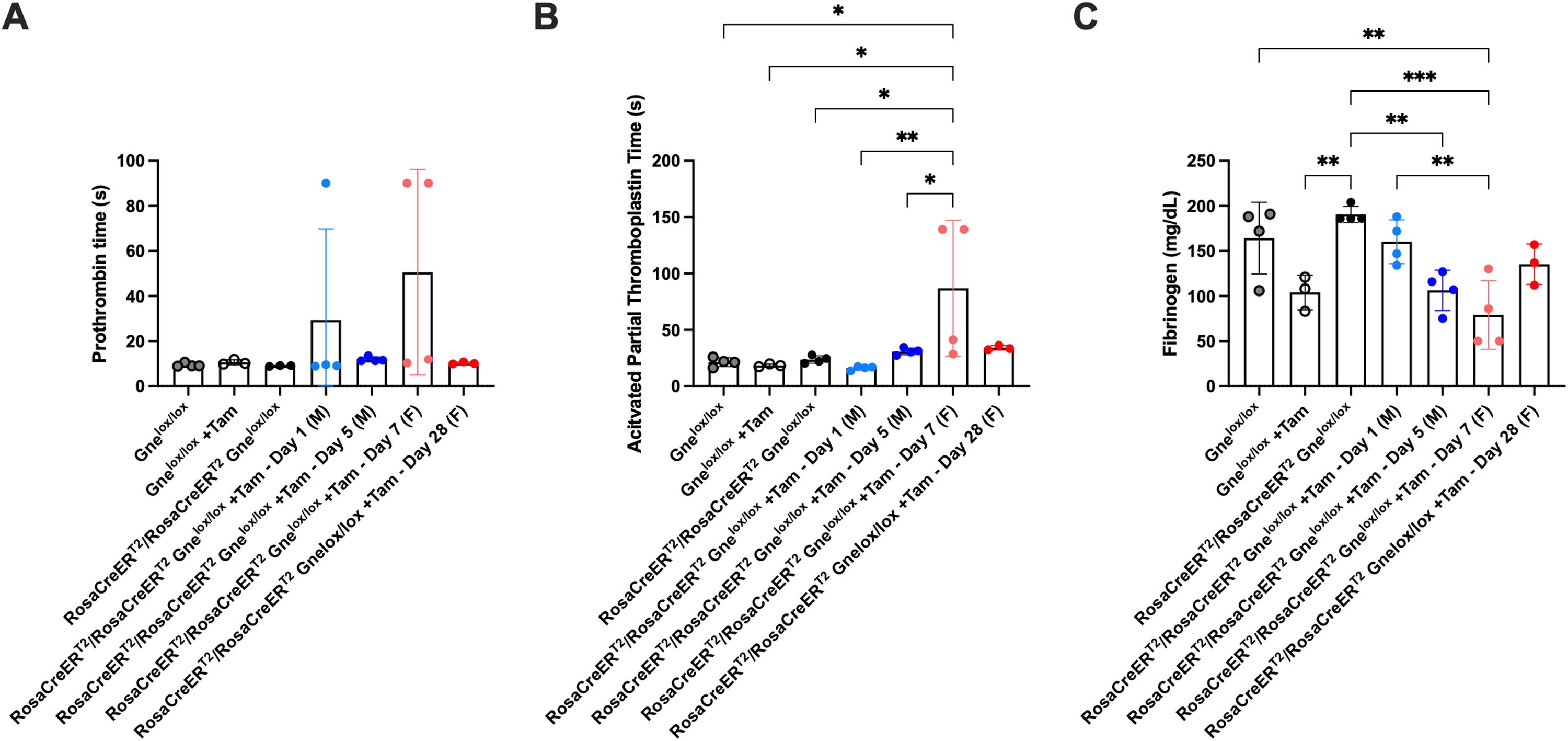
Variable changes in platelet clotting function with systemic Gne gene deletion in adult mice. Pro-thrombin time (A), activated partial thromboplastin time (B), and fibrinogen protein levels (C) were measured in plasma isolated from untreated and tamoxifen (+Tam)-treated Gnelox/lox and Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice. Treated mice were analyzed near endpoint (5 days for males and 28 days for females) or prior to endpoint (1 day for males and 7 days for females). Errors are SD. Statistical significance between groups determined by one-way ANOVA with Tukey's post-hoc test. *p < 0.05, **p < 0.01, ***p < 0.001. Untreated Gnelox/lox n = 4; Gnelox/lox + Tam n = 3; untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox n = 3 or 4; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day1 (M) n = 4; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day5 (M) n = 4; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day7 (F) n = 4; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox +Tam-Day28 (F) n = 3.
Giemsa staining of whole blood smears from tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox showed a near absence of platelets and an increase in nucleated RBCs and RBCs with abnormal membrane morphology (Figures 5A-D). Platelets were also reduced in stains of untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox blood relative to untreated HSA-CreERT2/+Gnelox/lox. Giemsa staining of bone marrow smears from untreated and tamoxifen-treated male Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice showed evidence of anemia at endpoint with tamoxifen treatment (Figures 5E-F). These findings included a near absence of RBCs with enhanced neutrophils and basophils. No significant changes were observed in either blood cell or bone marrow smears in tamoxifen-treated HSA-CreERT2/+Gnelox/lox or Rosa-CreERT2/Rosa-CreERT2 mice or in untreated Gnelox/lox mice (ns). Gne exon 3 was deleted (Figure S11A) and Gne gene expression was significantly reduced (Figure S11B), most often to 4–10% of normal, in bone marrow isolated from male and female tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice at endpoint.
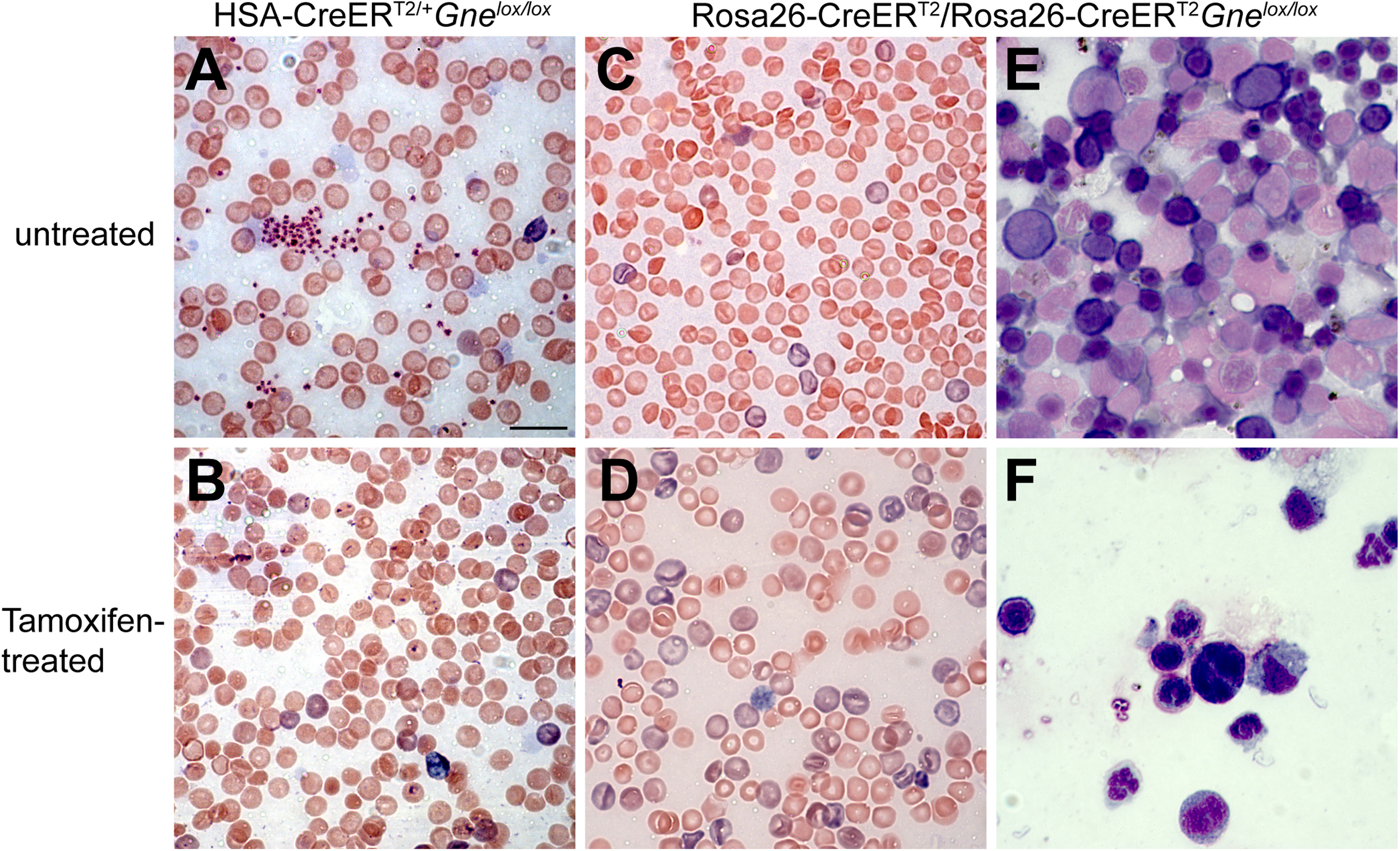
Giemsa staining of blood and bone marrow smears. Giemsa stained whole blood (A-D) and bone marrow (E-F) smears were taken from untreated (A, C, E) or tamoxifen-treated (B, D, F) HSA-CreERT2/+Gnelox/lox mice (A, B) or Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice (C-F). Blood and bone marrow smears show tamoxifen treatment at endpoint (B, D, F). Bar is 10 μm for all panels.
We observed increased staining in both platelets and RBCs with lectins that bind glycans where a capping Sia is absent (PNA, ECA, and RCA) using flow cytometry (Figure 6). For RBCs (Figure 6A), PNA, ECA, and RCA all showed very significant increases in cell binding at 28 days post-tamoxifen treatment in female Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice compared to untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox controls, while elevations in male mice at 5 days post-tamoxifen treatment were not significantly changed. Lectins that bind Sia directly, MAA and SNA, were increased in RBCs at 7 days post-tamoxifen of female mice, while for males, increased binding was seen for MAA but not for SNA. For platelets (Figure 6B), both PNA and ECA were increased in 28 day female and 5 day male post-tamoxifen Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice. RCA binding was elevated for both 1 day and 5 day male mice, but not in 7 day or 28 day female mice. As with RBCs, some increased binding of lectins that bind Sia was also observed in platelets; MAA binding was significantly increased in 5 day male, 7 day female, and 28 day female mice, but no significant changes were observed in SNA binding. These results demonstrate increased expression of Sia-deficient glycans in platelets and sometimes in RBCs of tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice, and these results share some similarities with lectin binding changes seen in platelets from human patients with GNE mutations causing thrombocytopenia.7,8
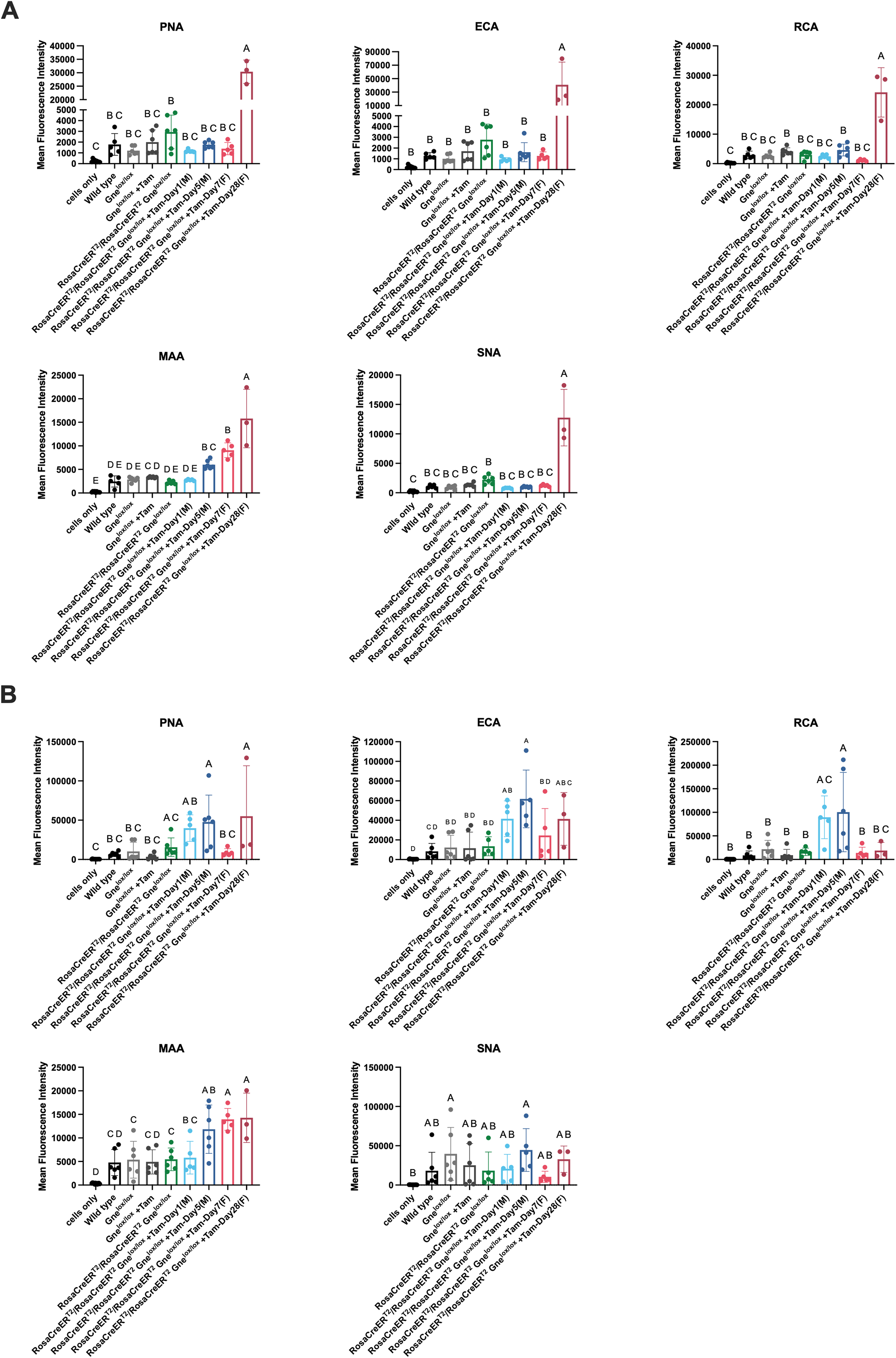
Increased expression of asialo-glycans in red blood cells and platelets after systemic Gne gene deletion. Lectins that bind asialo-glycans (PNA, ECA, RCA) or sialic acid (MAA or SNA) were used to stain red blood cells (A) or platelets (B). Cells were analyzed by flow cytometry. Tamoxifen (+Tam)-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox near lifespan endpoint (1 day for males and 7 days for females) or just prior to lifespan endpoint (5 day for males and 28 days for females) were compared to untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox, Gnelox/lox, and wild type mice as well as tamoxifen-treated Gnelox/lox mice. “Cells only” represents signal without lectin added. Errors are SD. Statistical significance between groups (p < 0.05) was determined by one-way ANOVA with Tukey's post-hoc test. Panel A: Cells only n = 9; Wild Type n = 5; Untreated Gnelox/lox n = 6; Gnelox/lox + Tam n = 6; untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox n = 5 or 6; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day1 (M) n = 5; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day5 (M) n = 6; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day7 (F) n = 5; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox +Tam-Day28 (F) n = 3. Panel B: Cells only n = 12; Wild Type n = 6; Untreated Gnelox/lox n = 6; Gnelox/lox + Tam n = 6; untreated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox n = 5 or 6; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day1 (M) n = 5; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day5 (M) n = 5 or 6; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox + Tam-Day7 (F) n = 5; Rosa-CreERT2/Rosa-CreERT2Gnelox/lox +Tam-Day28 (F) n = 3.
Deletion of Gne in skeletal myofibers does not cause myopathy, but systemic deletion in skeletal muscle does
While systemic deletion of exon 3 of Gne in Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice caused fatality with anemia, thrombocytopenia, and spontaneous bleeding, deletion of exon 3 of Gne specifically in skeletal myofibers in tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice led to none of these phenotypes. Complete blood cell counts showed only modest reductions in tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice relative to untreated controls (Table S3) at 60 days post-treatment, akin to what was seen with tamoxifen-treated Gnelox/lox mice (Figure 2A), and these reductions were not present at 270 days post-treatment (Table S3). Skeletal muscles in HSA-CreERT2/+Gnelox/lox mice also showed no evidence of muscle pathology at 60 or 270 days post-tamoxifen treatment (Figure 7); there was no increase in central myofiber nuclei, a sign of a cycle of muscle degeneration and regeneration (Figure 7B), despite a clear reduction in muscle Gne gene expression (Figure S3D, F), and myofibers did not significantly differ in size at either age, though a few treated mice did have reduced myofiber diameters at 60 days (Figures 7C, D). Affected skeletal muscles in tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice showed a dramatic increase in PNA staining at 60 days post-treatment, a lectin that binds Galβ−1,3-GalNAc- glycans only when Sia is absent, along skeletal myofibers, while ECA, a lectin that binds Galβ1,4GlcNAc only when Sia is absent, showed no change (Figure S12). Staining for lectins that bind Sia directly (MAA, SNA) was not obviously changed by tamoxifen treatment (Figure S13), and assay of total sialic acid amounts also showed no significant change (Figure S5B).
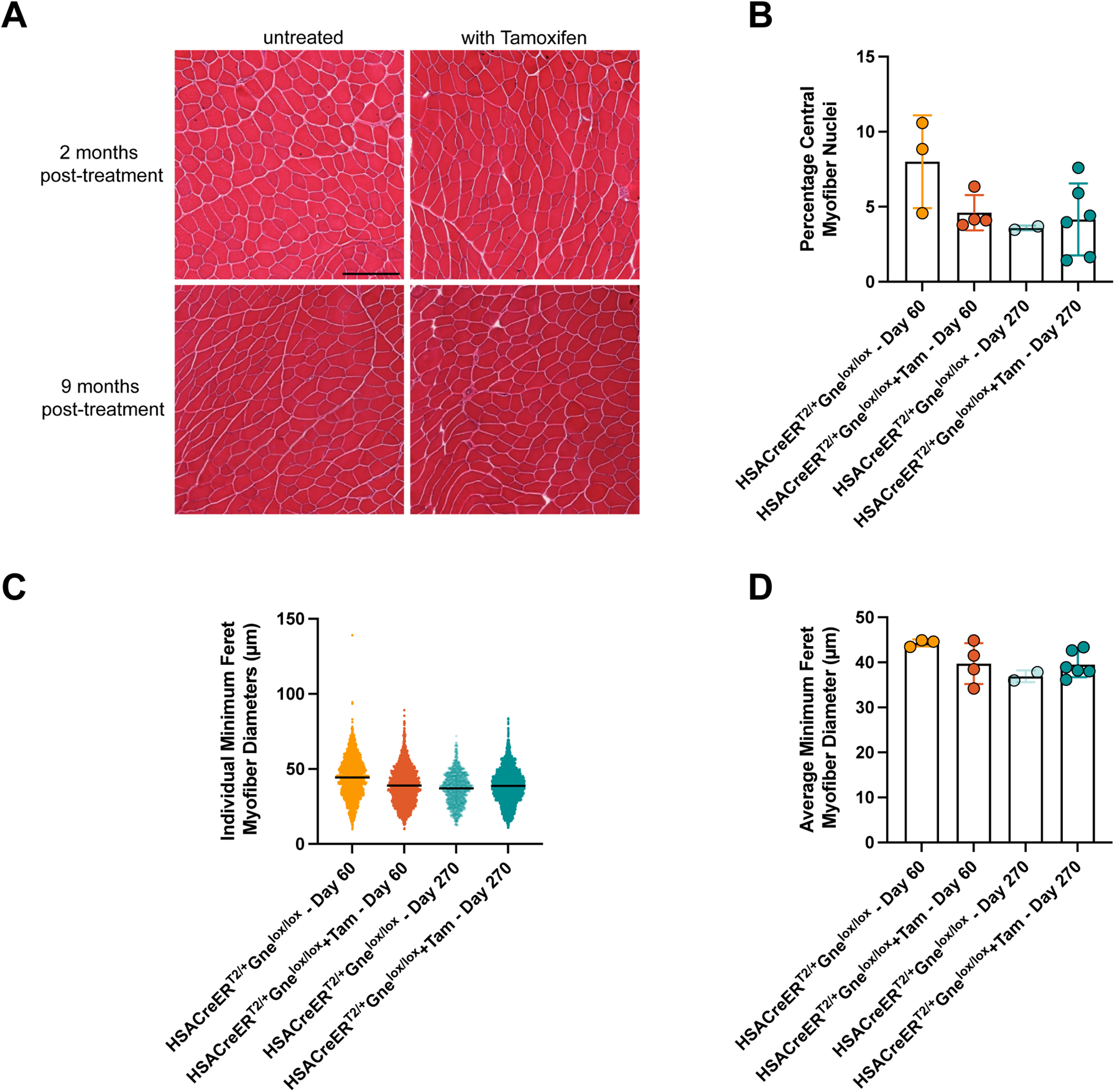
Absence of muscle pathology in tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice. (A) Hematoxylin and eosin-staining of tamoxifen (+Tam)-treated and untreated skeletal muscles at 60 days (2 months) and 270 days (9 months) post-treatment. Percentage of central myofiber nuclei (B), individual myofiber diameters (C), and average myofiber diameters per mouse (D) in muscles from tamoxifen-treated and untreated mice at 60 and at 270 days post-treatment. Bars is 200 μm for all panels in A. Errors are SD for B, C, and D. Statistical significance between groups determined by one-way ANOVA with Tukey's post-hoc test. Panel B and D: Untreated day 60 n = 3; +Tam Day 60 n = 4; Untreated Day 270 n = 2; +Tam Day 270 n = 6.
We next explored systemic deletion of exon 3 of the Gne gene in muscle using AAV injections (Figure 8) to express a functional GFP-Cre fusion protein, as we have done previously. 44 We performed biweekly intramuscular (IM) injections of AAVs bearing Cre so that it would be expressed only in skeletal myofibers using the MCK promoter, or systemically, using the CMV promoter. 4 week-old Gnelox/lox mice were injected in the tibialis anterior (TA), gastrocnemius (gastroc), and quadriceps femoris (quad) muscles once every 2 weeks for a total of 5 injections. Muscles were analyzed 2 weeks after the final injection (10 weeks after the first injection).
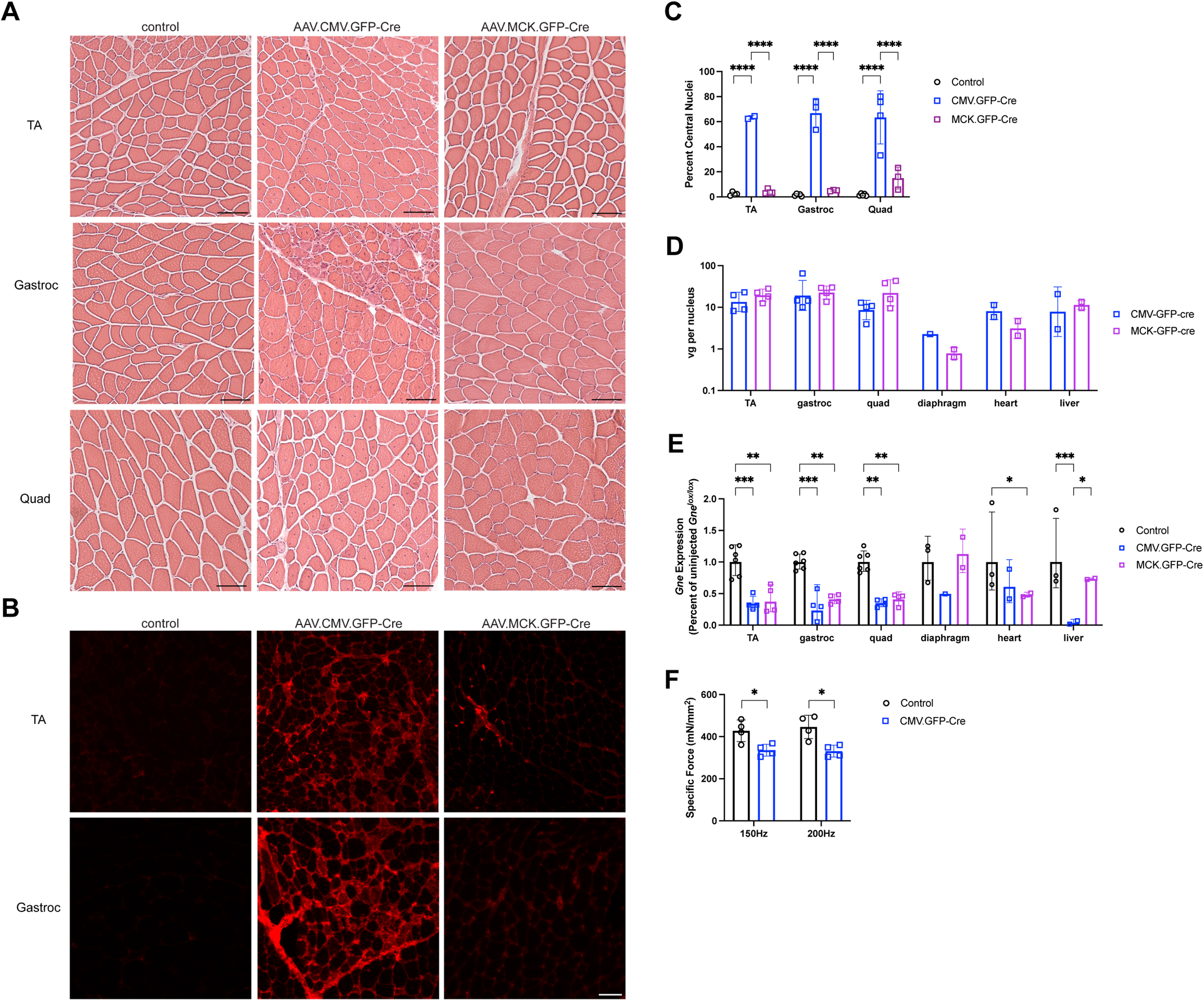
Presence of muscle pathology with systemic Gne gene deletion in Gnelox/lox muscles. Tibialis anterior (TA), gastrocnemius (Gastroc), and quadriceps femoris (Quad) muscles in Gnelox/lox mice were injected with AAV.CMV.GFP-Cre or AAV.MCK.GFP-Cre every two weeks for five total injections (A-E) or twice, one month apart (F), and compared to muscles from uninjected Gnelox/lox mice. Muscles were analyzed 2 (A-E) or 6 (F) weeks following the final injection, or 10 weeks after injections were started. (A) Hematoxylin and eosin staining. (B) Mouse IgG staining. (C) Percentage of central myofiber nuclei. (D) AAV biodistribution. vg = vector genomes. (E) Relative Gne gene expression, normalized to untreated Gnelox/lox control tissue set at 100%. (F) Maximal tetanic specific force in the TA measured in situ at 150 Hz or 200 Hz stimulation. Bar is 50μm in A, B. Errors are SD for C, D, E, F. Statistical significance between groups determined by one-way ANOVA with Tukey's post-hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant. Panel C: Control n = 4 or 5, AAV.CMV.GFP.Cre n = 2–4; AAV.MCK.GFP.Cre n = 3 or 4. Panel D: TA, Gastroc, and Quad n = 4; Diaphragm, Heart, and Liver n = 2 except AAV.CMV.GFP.Cre Diaphragm n = 1. Panel E: TA, Gastroc, and Quad control n = 6, injected n = 4; Diaphragm, Heart, and Liver control n = 3, injected n = 2 except diaphragm AAV.CMV.GFP.Cre n = 1. Panel F: n = 4 for all conditions.
Hematoxylin and eosin (H&E) staining of muscle sections showed muscle damage in a majority of myofibers after injection with AAV.CMV.GFP-Cre; increased percentages of myofibers with central nuclei indicated that most myofibers had undergone a cycle of muscle degeneration and regeneration when CMV was used (over 60% of myofibers in each muscle), while no such increases were seen when MCK was used as the promoter (Figures 8A, C). Similarly, AAV.CMV.GFP-Cre-injected muscles had evidence of split myofibers in all three muscles analyzed, as well as the presence of necrotic and small regenerating myofibers, particularly in the gastrocnemius muscle. Use of the CMV promoter also drove high elevations in mouse IgG staining, an indicator of muscle membrane damage, while MCK showed little or no change (Figure 8B). There was a significant drop in tetanic specific force in AAV.CMV.GFP-Cre-injected TA muscle, measured in situ, when either a 150 Hz or a 200 Hz tetanic stimulation was given (Figure 8F). For this experiment, only two AAV injections were given, spaced a month apart, with the endpoint at 20 weeks after the first AAV injection. This led to the same dramatic changes in muscle pathology and reduced muscle Gne gene expression as was seen when five injections of AAV were used (ns). No inclusion bodies were observed within skeletal myofibers, as evidenced by Congo Red staining, when either promoter was used (Figure S14 and ns). Here, brain from a 12-month-old APPSwePSEN1dE9 mouse, a model of Alzheimer's disease was used as a positive control for Congo Red staining.45,46
Injection of AAV.MCK.GFP-Cre or AAV.CMV.GFP-Cre yielded almost equivalent amounts of AAV vector genomes (vgs) in affected muscles (TA, gastroc, quad, diaphragm) and also showed nearly equivalent vgs in heart and liver (Figure 8D). Both MCK- and CMV-driven vectors reduced Gne gene expression by 70–80% in injected limb muscles (TA, gastroc, quad, Figure 8E). AAV.CMV.GFP-Cre also significantly reduced Gne gene expression in the liver, while AAV.MCK.GFP-Cre did not (Figure 8E). While not significant, a similar trend was also seen in the diaphragm muscle, while both promoters reduced expression in heart. Background autofluorescence in the GFP channel made it difficult to assess GFP-Cre protein fluorescence directly (ns). To get around GFP fluorescence issues, we used Cy3 to mark lectin staining and AlexaFluor 647 to mark laminin alpha 2 staining. Here, we pseudocolored AlexaFluor 647 signal to green to more clearly show staining overlap.
We assessed binding of lectins that bind sialic acid (MAA, SNA) and lectins that bind glycans only when not capped by sialic acid (PNA, ECA) to see if patterns would change in muscle, as has been shown, to variable degrees, in a number of previous studies.40,47–49 Use of CMV to drive Cre expression induced an elevation of PNA and ECA lectin staining in skeletal muscle sections, both in skeletal myofibers and in blood vessels (Figure S15), while staining for MAA and SNA was modestly reduced (Figure S16). When MCK was used as the promoter, PNA staining was highly elevated in skeletal myofibers, but not in blood vessels, while ECA staining was not changed (Figure S15). For MCK.GFP-Cre, SNA and MAA staining were not obviously reduced (Figure S16). Total sialic acid measures in injected muscles, however, trended toward reduced levels, with a 30–40% reduction in mean levels for both MCK and CMV promoter-driven AAVs compared to untreated Gnelox/lox controls (Figure S5C).
We tested AAV.Cre-injected muscles for changed gene expression of markers of skeletal muscle regeneration (Figure 9A), including quiescent satellite cells (Pax7, paired box 7), activated satellite cells (Myf5, myogenic factor 5), myoblasts and immature myotubes (Myod1, myoblast determination protein 1, and Myog, myogenin), regenerating skeletal myofibers (Myh3, myosin heavy chain 3), and mature skeletal myofibers (Dmd, dystrophin, muscular dystrophy) (Figure 9A). We also tested for markers of muscle cell senescence (Cdkn2a, cyclin-dependent kinase inhibitor 2a), muscle cell polarity (Pard6g, par-6 family cell polarity regulator gamma), muscle denervation (Chrna1, nicotinic acetylcholine receptor A1 and Musk, muscle-associated receptor tyrosine kinase), and muscle atrophy (Fbxo32, F-box protein 32, also Atrogin-1 and Trim63, Tripartite motif-containing 63, also MuRF1) (Figure 9B). Injection with AAV.CMV.GFP-Cre significantly elevated Myf5, Myog, and Myh3 expression, consistent with induction of muscle damage and regeneration, while treatment with AAV.MCK.GFP-Cre did not. Myh3 (Figure 9C) and Cdkn2a (Figure 9D) protein staining mirrored the changes seen in gene expression. AAV.CMV.GFP-Cre decreased expression of Pax7, though not significantly so, and increased expression of Cdkn2a, consistent with satellite cell loss and senescence. There was significantly elevated expression of markers for muscle denervation, and variable, though sometimes large, increases in markers for muscle atrophy. Injection of AAV.MCK.GFP-Cre did not significantly change any of these measures despite lowering Gne gene expression to the same degree seen with CMV (Figure 8E).
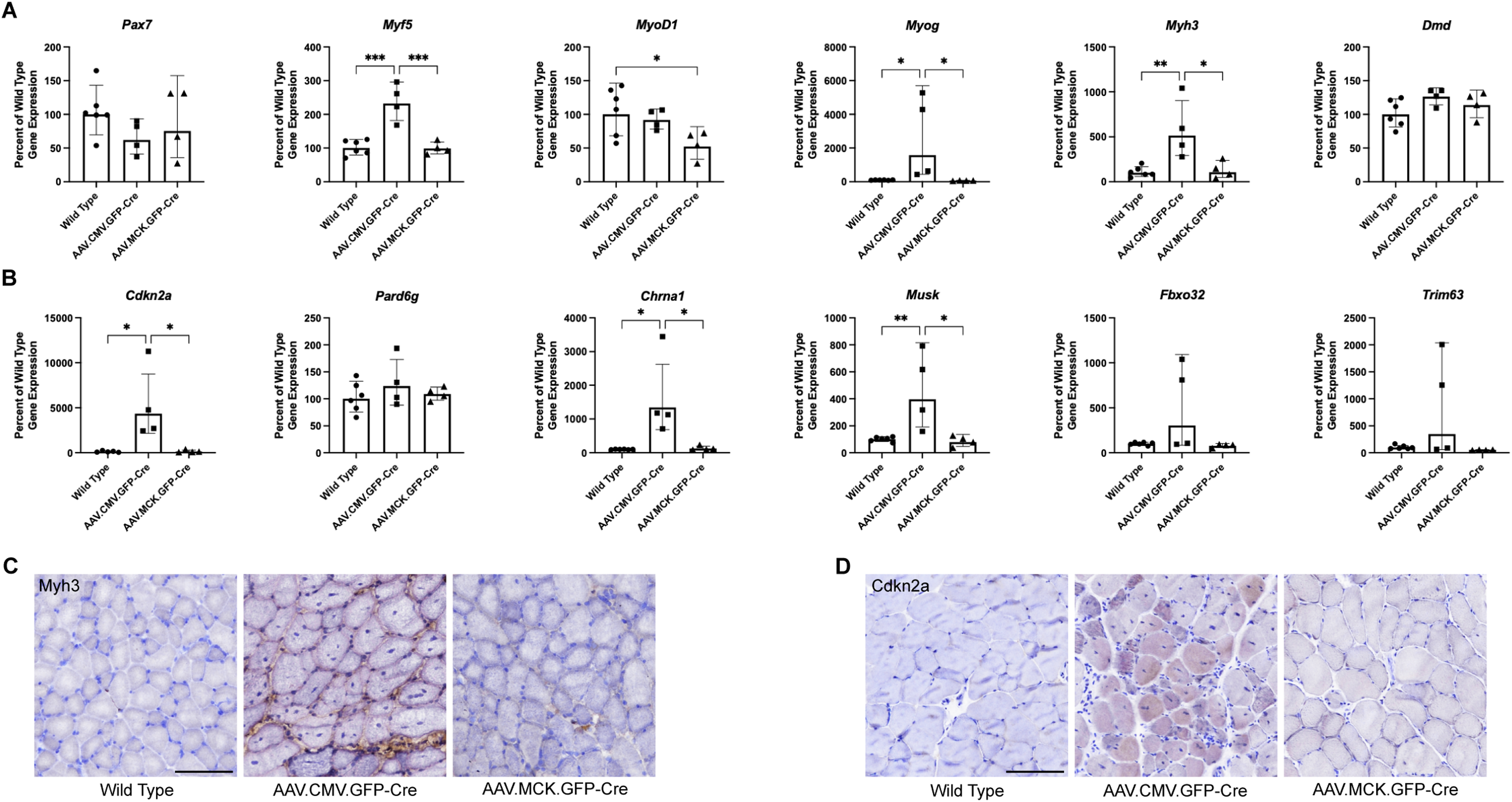
Expression of gene markers for muscle regeneration and pathology. Gene expression changes were measured by qRT-PCR in Gnelox/lox muscles injected with PBS (control, n = 6), AAV.CMV.GFP-Cre (n = 4), or AAV.MCK.GFP-Cre (n = 4). (A) Relative expression of muscle regeneration, including markers for quiescent satellite cells (Pax7), activated satellite cells (Myf5), myoblasts (Myod1, Myog), regenerating myofibers (Myh3), and mature myofiber (Dmd) was assessed. (B) Relative expression of markers for muscle cell senescence (Cdkn2a), muscle cell polarity (Pard6g), muscle denervation (Chrna1, Musk) and muscle atrophy (Fbxo32, Trim63) was assessed. Statistical significance between groups determined by one-way ANOVA with Tukey's post-hoc test. Immunostaining for Myh3 (C) or Cdkn2a(D) protein is shown. Bar is 100 μm.
Discussion
The GNE gene encodes the enzyme responsible for the committed steps in sialic acid (Sia) biosynthesis, and GNE and Sia are expressed in every tissue of the body, so it is surprising that recessive missense mutations in human GNE can give rise to seemingly tissue-specific diseases that occur during different periods of development, including infantile thrombocytopenia.7,8,26,50,51 juvenile ALS 13 or motor neuropathy, 15 and adult-onset myopathy. 16 Here, we have used an inducible gene deletion mouse model to begin to assess Gne gene function in the adult mouse. Systemic Gne deletion in adult mice caused fatality within one month after gene deletion, demonstrating that Gne is an essential gene in the adult. This is consistent with previous studies showing that embryonic deletion of Gne is results in loss of viability, 5 a result we have also confirmed here.
Death in tamoxifen-treated Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice was associated with spontaneous bleeding that likely resulted from mice having very low platelet counts. Patients with certain GNE mutations can develop severe thrombocytopenia, sometimes presenting in infancy.6–12,52 Thrombocytopenia can also co-present in adults with GNE myopathy, though levels of platelet reduction do not reach severe levels and spontaneous bleeding is not typically seen.26,28,50,52 Mice with embryonic deletion of Gne and mice bearing certain mutant Gne genes also develop fatal bleeding in the embryonic to neonatal period.53,54 Platelets lacking Sia can be more quickly cleared by the liver, 55 and this may explain the thrombocytopenia seen in induced Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice. Several mice showed slow platelet clotting times that could also have contributed to increased bleeding. The reduction in RBCs, hematocrit, hemoglobin, and WBCs with systemic Gne gene deletion could also result from bleeding related to thrombocytopenia. RBCs, however, may also be more quickly cleared when Sia is removed, through cellular or immune-mediated mechanisms. 56 The near absence of RBCs in bone marrow stains at endpoint suggests their production is reduced or eliminated after Gne gene deletion. The half-life of mouse RBCs is about 12 days, 57 so an endpoint of several weeks after gene deletion could account for the 50% reduction in RBCs that was seen in whole blood if new RBCs were not being made. In at least two subjects, RBCs were low despite platelet levels that were not low enough to induce spontaneous bleeding. The reduction in staining for RBCs, WBCs and megakaryocytes in bone marrow suggest their absence is caused by an aplastic anemia. It is interesting that while mutations in the human GNE are known to cause reductions in platelets, they have not been reported to cause anemia. It is possible that the level of gene deletion we have induced here is greater than that caused by GNE mutations associated with thrombocytopenia, leading to additional blood cell phenotypes.
Treatment of non-deleted Rosa-CreERT2/Rosa-CreERT2 mice as embryos or neonates with tamoxifen has been reported to induce severe anemia and bone marrow changes in the first few weeks after treatment.41,42 We controlled for this in our experiment, which was done on mature adult mice, and found only minor changes in red blood cell counts in mouse lines with no gene deletion given tamoxifen. Such reductions in red blood cells still led to total levels that remained within the normal range. The fact that we saw these same minor changes in tamoxifen-treated Gnelox/lox mice suggests such changes were not due to the use of the Rosa-CreERT2 transgenic line.
The severe lethal nature of systemic Gne gene deletion did not allow for a full assessment of reduced Gne gene expression in the nervous system or in skeletal muscle, systems impacted by GNE mutations in humans.2,13–17,58 To try to address the role of muscle more directly, we specifically deleted Gne in skeletal myofibers. We thought this approach would model disease found in patients with GNE myopathy, but deletion of Gne specifically in tamoxifen-treated HSA-CreERT2/+Gnelox/lox mice did not lead to any muscle disease for at least 9 months after gene deletion. This result confirms recently published work from Mitrani-Rosenbaum and colleagues showing that induced Gne deletion in skeletal myofibers and liver of adult mice gave rise to no disease for up to a year. 59 That same study showed that complete Gne gene deletion in skeletal myofibers, coupled with liver gene deletion, only partially reduced Sia levels in affected skeletal muscle. 59 We found little or no change in total Sia content in muscles where Gne was only deleted in skeletal myofibers, while a more systemic deletion did reduce muscle Sia levels. This result is consistent with other published findings suggesting that Sia can be loaded into muscle from other organs, for example liver 46 and therefore may require Gne gene deletion beyond skeletal myofibers to lower muscle Sia levels sufficiently to cause disease. Nevertheless, we did observe an increase muscle staining with lectins that recognize asialo glycans, akin to results seen by others in mouse models and in GNEM patients.40,47–49 This suggests a change in steady state Sia glycosylation of glycoproteins with muscle Gne gene deletion, even if total Sia levels are not significantly changed.
Systemic deletion of Gne using repeated intramuscular injection of AAV.CMV.GFP-Cre in Gnelox/lox mice caused significant myopathy at 10 weeks post injection. By contrast, equivalent injections of AAV.MCK.GFP-Cre did not, despite equivalently lowering overall muscle Gne gene expression. These experiments support the notion that cells in addition to skeletal myofibers must have Gne deleted in order for muscle pathology to occur. Injection of AAV.CMV.GFP-Cre increased expression of markers for muscle cell senescence, atrophy, regeneration, and denervation, and it lowered maximal muscle tetanic specific force. Such changes are consistent with mechanisms thought to occur in GNE myopathy, though aspects of muscle pathology found in this disease, such as the presence of inclusion bodies, were yet not seen at this early endpoint. Intramuscular satellite cells or motor neurons, each of which can be transduced to some degree by AAV after intramuscular injection,60,61 might need to be deleted in addition to skeletal myofibers in order for such pathologies to occur.
These inducible Gne gene deletion mice add to the repertoire of models for GNEM, which include previous transgenic (GNED207VTgGne−/−)62,63 and gene knock-in (GNEM743T/M743T) 43 models of human GNE gene mutations in mice. Each of these models provide strikingly different phenotypes, but they collectively contribute to a better understanding of the disease. GNED207VTgGne−/− mice can show muscle pathology and the presence of inclusion bodies in some muscles (e.g., tibialis anterior), but they must be aged up to a year to manifest such phenotypes and these muscle phenotypes can be variable in presentation and sometimes absent.62–64 Some GNEM743T/M743T mice perish from hematuria in the first two weeks of life, but others live extended lifespans without any phenotype being present.43,65 This is not unlike human patients with homozygous GNEM743T mutations, where disease presentation is variable66,67 and where several subjects have recently been reported with no apparent disease even in old age despite some of their siblings having GNEM. 68 What is clear from the current studies, and those of Mitrani-Rosenbaum and colleagues, 59 is that deletion of the Gne gene in adult mice can manifest some, but not all, GNEM disease phenotypes. Systemic deletion in Rosa-CreERT2/Rosa-CreERT2Gnelox/lox mice shows a clear requirement for the Gne gene in adult animals and the presence of severe thrombocytopenia, but this lethality precludes an assessment of muscle phenotypes, which take longer periods to develop. By contrast, systemic Gne gene deletion in skeletal muscle using intramuscular injection of AAV.CMV.GFP-Cre can induce muscle damage and loss of maximal muscle tetanic specific force, but these phenotypes present more as a dystrophy, with muscle inflammation present, than they do as an inclusion body myopathy. Again, such phenotypes may require longer (or shorter) time periods to manifest than were done in our study. Deletion only in skeletal myofibers, either locally with AAV.MCK.GFP-Cre in Gnelox/lox mice or in all muscles in HSA-CreERT2/+Gnelox/lox mice, suggests that deletion of muscle Gne gene alone does not cause muscle pathology. These inducible gene deletion lines, coupled with others currently being made, should provide the tools to more carefully investigate the mechanism by which GNEM and other GNE-dependent diseases occur. Such studies are needed to better understand how to develop effective treatments for patients with these disorders.
Methods
Sex as a biological variable
Both male and female mice were used in the study for each genotype. When gender led to significant differences in outcome, male and female mice were reported as separate groups.
Mice
Mice were housed in a barrier facility with 12 h light and 12 h dark cycles with access to food and water ad libitum (Cat # 2919, Teklad Global Rodent diet, Inotiv, West Lafayette, IN). Gne+/− and Gnelox/lox mice were made by the Mouse Biology Program at University of California Davis using CRISPR-Cas9 methods. Gne+/− mice (Gneem1(IMPC)Mbp), which contain deletion of exon 3 in the Gne gene, are available from the NIH Mutant Mouse Resource and Research Center (MMRRC) at UC Davis (stock #066718-UCD). Gnelox/lox mice were made by inserting loxP sites flanking exon 3 of the mouse Gne gene. Genomic DNA sequencing data describing the location of the loxP sites and the sequence remaining after Cre-mediated deletion are presented in Figure S1. Mice were bred into the C57Bl/6J background for multiple generations prior to use. Rosa26-CreERT2 (strain #008463, B6.129-Gt(ROSA26Sortm1(cre/ERT2)Tyj/J) and HSA-CreERT2 (strain #025750, Tg(ACTA1-cre/ESR1*)2Kesr/J) transgenic mice, both in the C57Bl/6J background, were purchased from Jackson Laboratories (Bar Harbor, ME) and bred to Gnelox/lox to generate Rosa-CreERT2/Rosa-CreERT2Gnelox/+, Rosa-CreERT2/Rosa-CreERT2Gnelox/lox and HSA-CreERT2/+Gnelox/lox mice.
Genotyping of mice
DNA was extracted from various muscles and organs using a DNeasy Blood and Tissue kit (Qiagen, Germantown, MD) as per manufacturer's protocol. Genotyping PCR was performed in a 20 μL reaction using OneTaq Hot Start 2× Master Mix (New England Biolabs, Ipswich, MA) as per manufacturer's protocol. Primers for genotyping (Gne-cKO-F 5′-CAGGGAAGGCTTTCTCTTGA-3′, Gne-cKO-R 5′- ACGGGACACCGTGTTCTTAT-3′ and Gne-cKO-mutR 5′- GCTTGTAAGCCAAACCTCTGACCC-3′) were obtained from Integrated DNA Technologies (Coralville, IA). All 3 primers were included in the PCR reaction. PCR conditions were 94 °C for 30 s followed by 30 cycles of 94 °C for 30 s, 58 °C for 30 s, and 68 °C for 1.5 min, with a final extension of 68 °C for 5 min. WT bands are 207 and sometimes also 1515 bp; the floxed band is 241 bp, and gene deletion post-Cre expression yields a band at 463 bp. DNA sequencing of genomic DNA regions containing loxP sites was done by Genewiz (Azenta Life Sciences, South Plainfield, NJ).
AAV production
All AAVs used were made and purified by Andelyn Biosciences (Columbus, OH). rss (recombinant single-stranded) AAVrh74.CMV.GFP-Cre and rssAAVrh74.MCK.GFP-Cre were produced by a triple-transfection method in HEK293 cells 69 and purified using iodixanol density centrifugation and anion exchange chromatography 70 and eluted in TMN200-P (200 mM NaCl, 1 mM MgCl2, 20 mM Tris, pH adjusted to 8.0 and supplemented with 0.001% Pluronic F-68). AAV titers were measured by qPCR against a supercoiled standard or through digital droplet PCR.
Tamoxifen and AAV administration to mice
Tamoxifen (Sigma-Aldrich, St Louis, MO) was dissolved in corn oil at a concentration of 20 mg/mL by incubating at 55° C with periodic vortexing to mix. Rosa-CreERT2/Rosa-CreERT2Gnelox/+, Rosa-CreERT2/Rosa-CreERT2Gnelox/lox and HSA-CreERT2/+Gnelox/lox mice, and strain controls, were administered with tamoxifen intraperitoneally at a dose of 75 mg/kg once daily for 5 consecutive days between 3 and 4 months of age. Gnelox/lox mice were given intramuscular (IM) injections of rssAAVrh74.CMV.GFP-Cre or rssAAVrh74.MCK.GFP-Cre into both left and right tibialis anterior, gastrocnemius and quadriceps muscles at doses of 1 × 1010 vg, 5 × 1011 vg, and 5 × 1011 vg, respectively. 4 week-old Gnelox/lox mice were injected IM with AAV, followed by four additional injections, given once every two weeks, after which a two week interval without injection occurred prior to analysis (at 10 weeks post the first injection). For muscle physiology studies, two AAV injections were given IM, spaced one month apart, with mice analyzed at 20 weeks post the first injection.
Hematology
Complete blood counts were performed on whole blood collected in EDTA tubes from mice via the submandibular vein or the retro-orbital vein using a Sysmex XP-300 Automated Hematology Analyzer (Sysmex America, Inc, Lincolnshire, IL). Citrated whole blood was collected from mice via the retro-orbital vein. Plasma was separated from citrated whole blood by centrifuging at 350×g for 20 min. Prothrombin time, activated partial thromboplastin time, and fibrinogen were measured from plasma by Antech GLP (Morrisville, NC).
Flow cytometry
Citrated whole blood was collected from mice via the retro-orbital vein. Platelet rich plasma (PRP) was collected by centrifuging citrated whole blood for 20 min at 350×g. Red blood cells were stained and gated with TER-119-BV421 (#116234; BioLegend, San Diego, CA). Platelets were stained and gated with CD41-BV421 (#133912; BioLegend, San Diego, CA). α−2,6- and α−2,3-linked sialic acid was detected using 20 μg/mL Cy5-labeled Sambucus nigra lectin (SNA) (#CL-1305; Vector Laboratories, Newark, CA) or 20 μg/mL Cy5-labeled Maackia amurensis lectin II (MAL II) (#21511110-1; GlycoMatrix, Dublin, OH), respectively. Surface galactose was stained with 10 μg/mL fluorescein isothiocyanate (FITC)–conjugated Ricinus communis I (RCA-1) (#FL-1081-1; Vector Laboratories, Newark, CA), 10 μg/mL FITC-conjugated Erythrina cristagalli (ECA) (#FL-1141-5; Vector Laboratories, Newark, CA), or 10 μg/mL FITC-conjugated peanut agglutinin (PNA) (#FL-1071-5; Vector Laboratories, Newark, CA). Cells were stained for 30 min at room temperature, then diluted in Tyrode's buffer without calcium before being acquired on a BD Fortessa flow cytometer (BD Life Sciences, Franklin Lakes, NJ), with at least 10,000 gated events recorded. Data were analyzed using FlowJo software, version 10.8.2 (BD Life Sciences, Franklin Lakes, NJ).
AAV biodistribution
DNA was extracted from various muscles and organs using a DNeasy Blood and Tissue kit (Qiagen, Germantown, MD) as per manufacturer's protocol. DNA concentration was measured on a nanodrop (Thermo Fisher Scientific, Waltham, MA) and diluted to 20 ng/μL. 100 ng of DNA was used to assess AAV vector genome (vg) copy number by quantitative PCR using an ABI7500 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA). Primers/probes designed for detecting CMV.GFP-Cre or MCK.GFP-Cre (5′-GAACCGCATCGAGCTGAA-3′, 5′- TGCTTGTCGGCCATGATATAG-3′, 5′-/56-FAM/ATCGACTTC/ZEN/AAGGAGGACGGCAAC/3IABkFQ/-3′) were obtained from Integrated DNA Technologies (Coralville, IA). Linearized plasmid DNA was used to create a standard curve of serial dilutions ranging from 10–107 copies/μL.
RT-qPCR
Total RNA was isolated using a RNeasy Mini kit (Qiagen, Germantown, MD), or a Direct-zol RNA miniprep kit (Zymo Research, Irvine, CA) as per manufacturer's protocol. RNA concentration was measured using a nanodrop (Thermo Fisher Scientific, Waltham, MA) and 500 ng of total RNA was used for first strand cDNA synthesis using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA). Primer/probe set for mouse Gne (Assay ID Mm.PT.58.42080332) targeting exons 3–4 was obtained from Integrated DNA Technologies (Coralville, IA). Primer probe sets for human GNE (Hs01103400_m1; exons 5–6), mouse Pax7 (Mm00834079_m1), mouse Myf5 (Mm00435125_m1), mouse Myod1 (Mm00440387_m1), mouse Myog (Mm00446194_m1), mouse Myh3 (Mm01332463_m1), mouse Dmd (Mm01216926_m1), mouse Cdkn2a (Mm00494449_m1), mouse Musk (Mm01346929_m1), mouse Chrna1 (Mm00431629_m1), mouse Fbxo32 (Mm00499523_m1), and mouse Trim63 (Mm01185221_m1) were obtained from Thermo Fisher Scientific (Waltham, MA). RT-qPCR assays were measured on an ABI7500 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA), with results analyzed using the delta-delta CT method 71 normalized to mouse 18S ribosomal (r)RNA (Cat# 4319413E; Thermo Fisher Scientific, Waltham, MA).
Western blotting
Tissue (liver or kidney) was homogenized in NP40 lysis buffer (TBS pH 7.4, 1% v/v NP-40, 0.5 mM EDTA and Protease Inhibitor (Roche, Indianapolis, IN)). Homogenized tissue was centrifuged at 10,000×g for 10 min at 4 °C and the supernatant was transferred to a new tube. Protein concentration was measured using a Bradford assay (Thermo Fisher Scientific, Waltham, MA). 40μg of protein was loaded on a 4–15% TGX Stain Free gel (Bio-Rad, Hercules CA), and then transferred to LF-PVDF membrane. After transfer, the membrane was blocked for 1 h at room temperature with Intercept Blocking Buffer (LICORbio, Lincoln, NE). The membrane was probed overnight at 4 °C with antibodies to GNE (1:2000 rabbit anti-GNE, 25079-1-AP, ProteinTech, Rosemont, IL) and GAPDH (1:10,000 mouse anti-GAPDH, G8795, Sigma-Aldrich, St Louis, MO) in Intercept Blocking Buffer with 0.01% (v/v) Tween-20. The membrane was washed 5× for 5 min each with TBST, then probed with secondary antibody (1:20,000 IRDye 680RD Goat anti-Mouse IgM (µ chain specific) and 1:20,000 IRDye 800 Goat anti-rabbit IgG, LICORbio, Licoln NE) for 1 h at room temperature. The membrane was again washed 5× for 5 min each with TBST before being imaged on a LICORbio Odyssey CLx imaging system.
Sialic acid quantification
Sialic acids were quantified by the periodate/resorcinol method. 72 Briefly, gastrocnemius muscles were weighed and homogenized in PBS to 0.05 mg/μl. 1 mg of muscle lysate was oxidized by the addition of 10 μl of 0.032 M periodic acid followed by incubation for 1.5 h at 37 °C to quantitate bound SA or at 4 °C to quantitate total SA content, followed by adding 100 μl of resorcinol reagent (0.6% (w/v) resorcinol, 2.5 μM CuSO4, 45% (v/v) concentrated HCl) and incubating at 80 °C for 1 h. After cooling, 100 μl of 95% t-butyl alcohol was added to each sample and absorbance was measured at 630 nm on a Spectramax ID5 plate reader (Molecular Devices, San Jose, CA). A standard curve was generated from known concentrations (50, 25, 12.5, 6.25, 3.125, 1.565, 0 nmol) of N-Acetyl neuraminic acid (NANA; TRC Canada, Vaughan, Canada) and the sialic acid concentrations were calculated by comparison with the standard curve.
Histology
Harvested non-muscle organs (liver, spleen, kidney, colon, lungs, and heart) were immersed in OCT before being frozen in dry ice-cooled isopentane. Skeletal muscles (diaphragm, tibialis anterior, gastrocnemius, quadriceps femoris, and triceps brachii) were mounted in OCT on corks and snap frozen in liquid nitrogen-cooled isopentane. 10 μm frozen tissue sections were prepared on a cryostat (Leica CM1950). Hematoxylin & eosin (H&E) staining was performed by first fixing slides in 10% neutral buffered formalin (Thermo Fisher Scientific, Waltham, MA) for 2 min. Fixed slides were then briefly washed with cold water, immersed in hematoxylin (Thermo Fisher Scientific, Waltham, MA) for 1.5 min, rinsed in warm tap water, then submerged in Shandon bluing reagent (Thermo Fisher Scientific, Waltham, MA) for 30 s. After rinsing with water, slides were dipped in Eosin-Y (Thermo Fisher Scientific, Waltham, MA) for 30 s, dehydrated with a series of 90%, 95% and 100% ethanol and cleared with several rinses in xylene. Slides were then mounted in Cytoseal XYL mounting media (Thermo Fisher Scientific, Waltham, MA) and covered with a coverslip. Congo Red staining was performed using an Alkaline Congo Red Amyloid Stain Kit (Poly Scientific R&D Corp, Bay Shore, NY) as per manufacturer's protocol. Giemsa staining was performed on blood and bone marrow smears using Giemsa stock stain (Newcomer Supply, Middleton, WI) as per manufacturer's protocol. All slides were imaged on a Zeiss Axioskop 40 microscope at 10x, 20x, or 100× magnification using an Axiocam 305 color camera using Zen blue v.3.4 software (Zeiss, Jena, Germany) or on a Precipoint M8 microscope and slide scanner at 20× magnification using MicroPoint software (PreciPoint GmbH, Munich, Germany).
Immunostaining
For fluorescence staining of mouse IgG,10 μm sections of skeletal muscles were blocked in 10% goat serum in 1× PBS overnight at 4 °C. Cy3-conjugated goat anti-mouse IgG antibody (Jackson ImmunoResearch; West Grove, PA) was added to the sections and incubated at room temperature for 1 h. Sections were washed in PBS and mounted using ProLong Gold antifade mountant with DAPI (ThermoFisher Scientific; Waltham, MA) and visualized using the Nikon Ti2-E widefield fluorescence microscope. For lectin staining, 10 μm sections of skeletal muscle were blocked in 2% fish gelatin in 1X PBS for an hour at room temperature. Biotinylated peanut agglutinin (PNA, B-1075-5), erythrina cristagalli lectin (ECA, B-1145-5), sambucus nigra lectin (SNA, B-1305-2), and maackia amurensis lectin II (MAA, B-1265-1) (all from Vector Laboratories; Plain City, OH) were added to the appropriate muscle sections along with rat monoclonal anti-laminin 2, α2 chain (L0663; Sigma Aldrich; St Louis, MO) and incubated overnight at 4 °C. Cy3-conjugated streptavidin (Jackson ImmunoResearch; West Grove, PA) and AlexaFluor 647-conjugated anti-rat IgG (Invitrogen; Waltham, MA) were added for an hour at room temperature, then slides were coverslipped using ProLong Gold antifade mountant with DAPI (ThermoFisher Scientific; Waltham, MA) and protected from light until imaging. Images were captured using the Nikon Ti2-E widefield fluorescence microscope, at which time the AlexaFluor 647 channel was pseudocolored to green for ease of visualization compared to red. For staining to quantify myofiber central nuclei and myofiber size, 10 μm sections of skeletal muscle were blocked with 10% goat serum in 1X PBS for 1 h at room temperature. Recombinant rabbit anti-dystrophin antibody (ab154168; Abcam; Waltham, MA) was added to the sections and incubated at 4 °C overnight. AlexaFluor 555-conjugated anti-rabbit IgG (Invitrogen; Waltham, MA) was added for an hour at room temperature, then slides were coverslipped using ProLong Gold antifade mountant with DAPI (ThermoFisher Scientific; Waltham, MA) and protected from light until imaging. Images were captured for analysis using the Nikon Ti2-E widefield fluorescence microscope with a Lumencor SOLA LED engine and Hamamatsu ORCA Fusion camera with a Nikon Plan Apochromat Lambda 10× objective. The final image pixel resolution was 0.64 μm/px. Identical exposures and other imaging settings were used for capturing all images. Non-overlapping images were taken to account for the entire muscle section in each mouse studied. Image quantification of myofiber diameter and percent central nuclei was performed using a custom automated analysis workflow developed in NIS-Elements AR software using the General Analysis 3 software module (Nikon; Melville, NY) and were quantified as previously described.73,74
For immunohistochemical staining, 10 μm muscle sections were cut on a cryostat (Leica CM1950). For embryonic myosin (Myh3) staining, sections were blocked using the M.O.M. (Mouse on Mouse) ImmPRESS HRP polymer kit (Vector Laboratories, Newark, CA) as per manufacturer's protocol. Primary antibody was used at 1:10 dilution (MHCD, Leica Biosystems, Deer Park, IL). For Cdkn2a staining, sections were fixed in 4% (v/v) paraformaldehyde in PBS, washed 3× in PBS, and blocked in 2.5% goat serum. Primary antibody (1:500, ab211542, abcam, Cambridge, MA) was incubated overnight at 4 °C, washed 3× with PBS, then incubated with ImmPRESS goat anti-rabbit HRP (Vector Laboratories, Newark, CA) for 30 min at room temperature. All slides were stained with ImmPACT DAB substrate (Vector Laboratories, Newark, CA) as per manufacturer's protocol, counterstained with Mayer's hematoxylin, dehydrated in ethanol and cleared in xylene before being mounted in Cytoseal XYL mounting media (Thermo Fisher Scientific, Waltham, MA).
Muscle physiology
Maximal specific tetanic force was measured in the tibialis anterior muscle in situ as previously described in Lam et al. 73
Statistics
Statistical analyses were performed using GraphPad Prism 10.4 software (GraphPad Software, La Jolla, CA). Unpaired two-tailed t tests or one way analysis of variance (ANOVA) with post hoc comparisons using Tukey's or Dunnett's multiple comparison test were performed. For platelet analysis, one sample t test with post hoc Wilcoxon test was also performed. p < 0.05 was considered statically significant in all tests. Kaplan–Meier curves were used to estimate survival with log-rank (Mantel-Cox) test used to determine significance.
Study approval
All mice were kept and studied in accordance with protocols approved by the Institutional Animal Care and Use Committee at the Abigail Wexner Research Institute at Nationwide Children's Hospital (Columbus, OH).
Supplemental Material
sj-pdf-1-jnd-10.1177_22143602251405918 - Supplemental material for Gne deletion in adult mice can cause thrombocytopenia, anemia, myopathy, bleeding, and death
Supplemental material, sj-pdf-1-jnd-10.1177_22143602251405918 for Gne deletion in adult mice can cause thrombocytopenia, anemia, myopathy, bleeding, and death by Patricia Lam, Deborah A Zygmunt, Macey Bennett, Anna Ashbrook, Jessica Hefty and Paul T Martin in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
We would like to thank the Mouse Biology Program at UC Davis for creating, by contract, inducible and Gne gene deletion mice, and the NIH Mutant Mouse Resource and Research Center (MMRRC) for funding assistance in making Gne gene deletion mice. We would like to thank Andelyn (Columbus, OH) for making adeno-associated virus reagents used in this study. Funding was supported by a grant from the Neuromuscular Disease Foundation (Beverly Hills, CA), a Technology Development Award from the Office of Technology and Commercialization at Nationwide Children's Hospital, and the National Institutes of Health (grant P50 HD117373).
Author contributions
PL, DAZ, and PTM were responsible for experimental design, manuscript writing, and most manuscript editing. PL performed flow cytometry, CBC analysis, histochemistry, and molecular studies. DAZ performed mouse genetics and pathology, AAV and tamoxifen injections, immunofluorescence, histochemistry, and physiology studies, AA performed AAV injections and whole animal analysis, MB performed molecular studies, and JH performed histochemistry studies. PTM was responsible for conceptualization and for obtaining funding support.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Nationwide Children's Hospital, Neuromuscular Disease Foundation, Eunice Kennedy Shriver National Institute of Child Health and Human Development, (grant number P50 HD117373).
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: PTM and Nationwide Children's Hospital have a financial conflict of interest with Genosera, Inc.
Data availability
All data are shown in the manuscript unless denoted (ns), not shown. Those additional study data may be requested from the authors through e-mail communication.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.