
Abstract
Introduction
The Myotonic Dystrophy Family Registry (MDFR) is a disease-specific, patient-reported online registry designed to capture real-world data on disease burden and lived experience in myotonic dystrophy (DM). By integrating patient-reported data with mechanisms for participant engagement, the MDFR provides a platform to characterize disease heterogeneity and support clinical research and trial readiness.
Aims
To characterize patient-reported disease burden and registry participation in DM, and to explore associations between disease subtype, age-related variables, and repeat expansion length.
Methods
De-identified MDFR data collected between 2013 and 2024 were aggregated and analyzed. Patient demographics and symptom prevalence were tested for associations between DM subtype, age, and repeat expansion length.
Results
The analysis demonstrated substantial, age-dependent disease burden in both DM1 and DM2, with multisystem involvement across central nervous system, cardiac, pulmonary, visual, and neuromuscular domains. Daytime sleepiness, myotonia, and fatigue were among the most frequently reported manifestations. Significant genotype–phenotype associations were identified in DM1, with CTG repeat length associated with increased prevalence of motor, cardiac, and central nervous system manifestations.
Conclusions
Patient-reported data from the MDFR provide complementary insights into disease burden and heterogeneity in DM, including domains that may be underrepresented in clinically derived datasets. Going forward, these findings support the integration of patient-reported, clinical, and genetic data to improve disease characterization, inform clinical trial design, and advance therapeutic development. Continued efforts to harmonize and expand longitudinal data collection and standardize data elements will be important to enhance the utility of registries.
Keywords
Introduction
Myotonic dystrophy (DM) refers to a group of multisystem, genetically defined disorders characterized by marked heterogeneity in symptom type, severity, and age of onset.1–3 This variability complicates diagnosis and presents challenges for clinical trial design, including endpoint selection, patient stratification, and interpretation of outcomes. 4 Persistent gaps in the characterization of disease burden and progression further limit the ability to define clinically meaningful outcomes and optimize therapeutic development.5,6
Patient registries provide a complementary approach to characterizing disease burden in rare disorders such as DM, where traditional clinical datasets may be limited in scale or scope.7,8 In contrast to clinician-driven natural history studies, patient-reported registries capture aspects of disease burden and lived experience that are not fully represented in clinically curated datasets. Compared with natural history studies, registries are typically less restrictive in design and enrollment, enabling broader inclusion and real-world representation, but often with reduced clinical standardization. These data extend structured clinical assessments by providing broader insight into symptom variability and real-world impact, while also supporting trial readiness, participant identification, and research engagement. 9
The Myotonic Dystrophy Family Registry (MDFR), established in 2013, is a disease-specific, patient-reported, online registry designed to capture information on disease manifestations, patient experience, and the impact of DM from the perspective of affected individuals and families. 10 In addition to its role as a data resource, the MDFR was designed in part as a contact registry to facilitate communication with participants regarding research opportunities, including clinical studies. By combining large-scale patient-reported data collection with mechanisms for participant engagement, the MDFR provides a unique platform to examine patterns of disease burden across subgroups and to support clinical research.
DM comprises two genetically distinct subtypes: type 1 (DM1), caused by a CTG repeat expansion in the DMPK gene on chromosome 19, and type 2 (DM2), caused by a CCTG repeat expansion in the CNBP (ZNF9) gene on chromosome 3. DM1 is typically associated with distal muscle involvement and may present as a congenital form with developmental impairment, whereas DM2 more often involves proximal muscles, with later onset and generally slower progression. 11 Both subtypes are progressive disorders without approved disease-modifying therapies, despite an increasingly active therapeutic development landscape.1,2
In this study, we performed a comprehensive analysis of de-identified MDFR data collected between 2013 and 2024. We characterized registry participation, patient demographics, and patient-reported symptom burden, and explored associations between DM subtype, age-related variables, and repeat expansion length. Our objective was to assess the extent to which patient-reported registry data can provide meaningful insights into disease burden and heterogeneity, and to evaluate their potential to complement existing datasets in support of clinical research and therapeutic development.
Methods
Study design
The MDFR launched in 2013 as a tool to help researchers and the myotonic dystrophy community learn more about the impact of this disease, and to organize the DM patient community for studies and trials. It is a patient-reported, online survey, annually approved by an Institutional Review Board. It operates under the guidance of a Principal Investigator and, a fully dedicated Registry Coordinator oversees registry data, activities and performance (Supplementary Information 1: Myotonic Dystrophy Family Registry). It is offered in English language only and available worldwide to DM1 and DM2 patients, of all ages. Children, younger than 18 years of age, or adults who cannot make their own medical decisions, must have their legal guardian, parent, or custodian register on their behalf.
The MDFR consists of a series of surveys organized in three sections 12 (Supplementary Information 2: Myotonic Dystrophy Family Registry Survey). Each section captures the core data elements that were established at the “Patient Registries and Trial Readiness in Myotonic Dystrophy” workshop. 6 Section 1 focuses on diagnosis, genetic test results, and repeat length. Section 2 contains questions relating to common symptoms as well as treatments and strategies utilized by participants to manage such symptoms. Section 3 captures information regarding the patients’ quality of life and includes questions pertaining to financial impact of the disease, employment status and living conditions. The current study focuses on results from Section 1 and Section 2.
Data on symptom prevalence as well as symptom impact/burden are captured in the Registry.
Symptom prevalence: For each symptom queried, a “yes”, “no” or “I don’t know” response is available (symptom prevalence).
Symptom burden: A 3-point scale system (0 = no effect, 1 = yes, mildly, and 2 = yes, severely) is used to record if a particular symptom has a negative effect on patients’ daily activities or on enjoyment of life (Supplementary Table 1: Symptom Burden, 3-point scale system). Only responses within the “Severe effect” category (Point 2) were captured for this study, analyzed, and reported. For surveyed symptom “Pain”, response options “quite a bit” and “very much” were collapsed into the Point 2 category (Severe effect) (Supplementary Table 1).
Study participants
The present study analyzed de-identified data from DM1 and DM2 patients, who joined the MDFR between February 2013 and July 2024. Registry participants were selected for inclusion in this study according to the following criteria:
DM1 subtypes were defined according to the reported age of when DM symptoms first manifested: from birth to 1 year of age, Congenital Myotonic Dystrophy (CDM) subtype; from 1 year of age to 18 years of age, Juvenile Onset subtype; 18 years of age and older, Adult DM1 subtype.
MDFR participants who did not report a DM diagnosis and age of symptom onset, and DM1 participants whose reported DM1 subtype/age of symptom onset did not align with the definition above, were excluded from the analysis.
Statistics
Data analyses were performed using R software, version 4.2. Descriptive statistics were used to summarize symptom prevalence in participants with DM1 and DM2. Results are reported as means ± standard deviations or as percentages, as appropriate.
Logistic regression was used to evaluate associations between CTG repeat length and clinical manifestations in adult DM1 participants (Subset 2, Table 5).
After excluding gender as a predictive variable, secondary analyses were performed using analysis of variance (ANOVA) and Student’s t-test to evaluate group differences by age (Table 4). Statistical significance was defined as a two-sided p-value <0.05.
Due to small sample sizes, the associations between age and symptom occurrence, and between CTG repeat length and symptom occurrence, were not examined in CDM or Juvenile participants.
Results
Registry cohort composition by inclusion criteria
Registry participants represented in study groups Subset 1 and Subset 2.

*Represents 63% of total Registry participants.
**Represents 23.5% of total Registry participants.
Adult-onset DM1 was the most prevalent category in both subsets, while congenital DM1 (CDM), juvenile-onset DM1, and DM2 were comparatively underrepresented.
Participants and subgroup demographics
Patient demographics study group Subset 1.
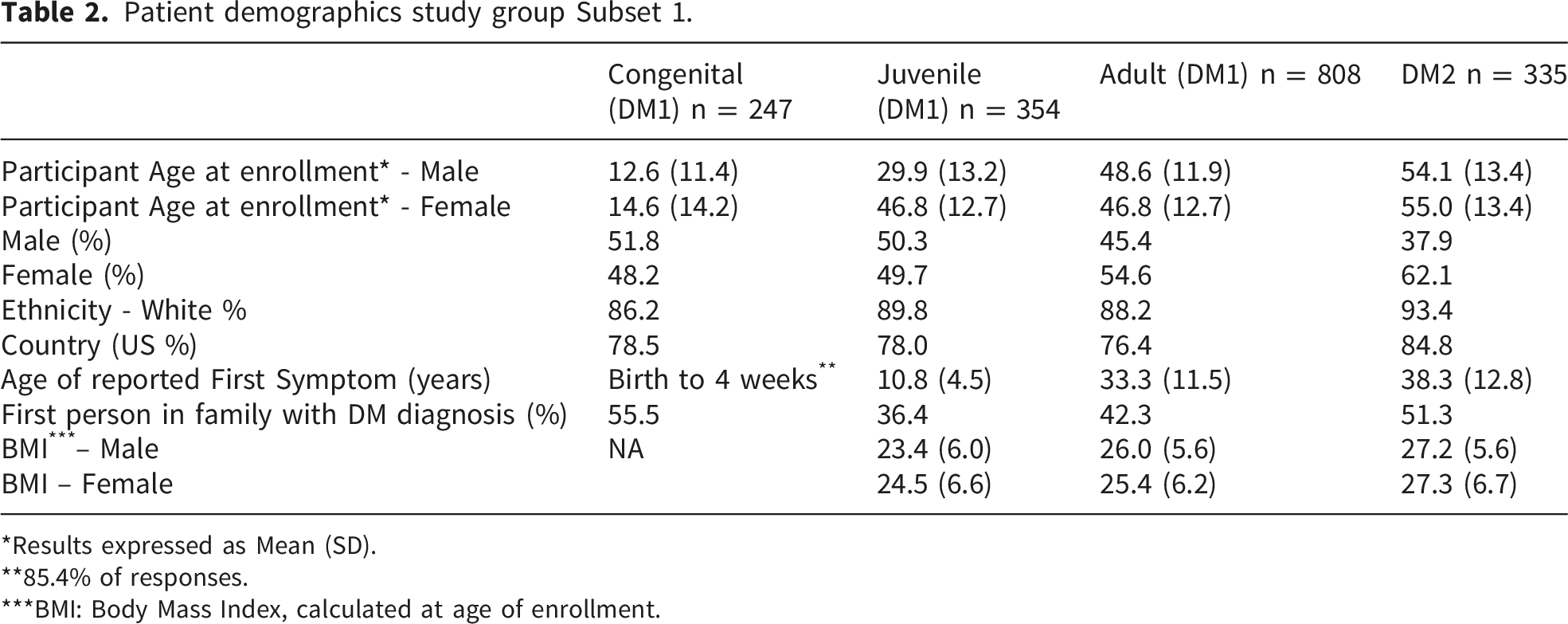
*Results expressed as Mean (SD).
**85.4% of responses.
***BMI: Body Mass Index, calculated at age of enrollment.
Demographics by disease type in study group subset 2.
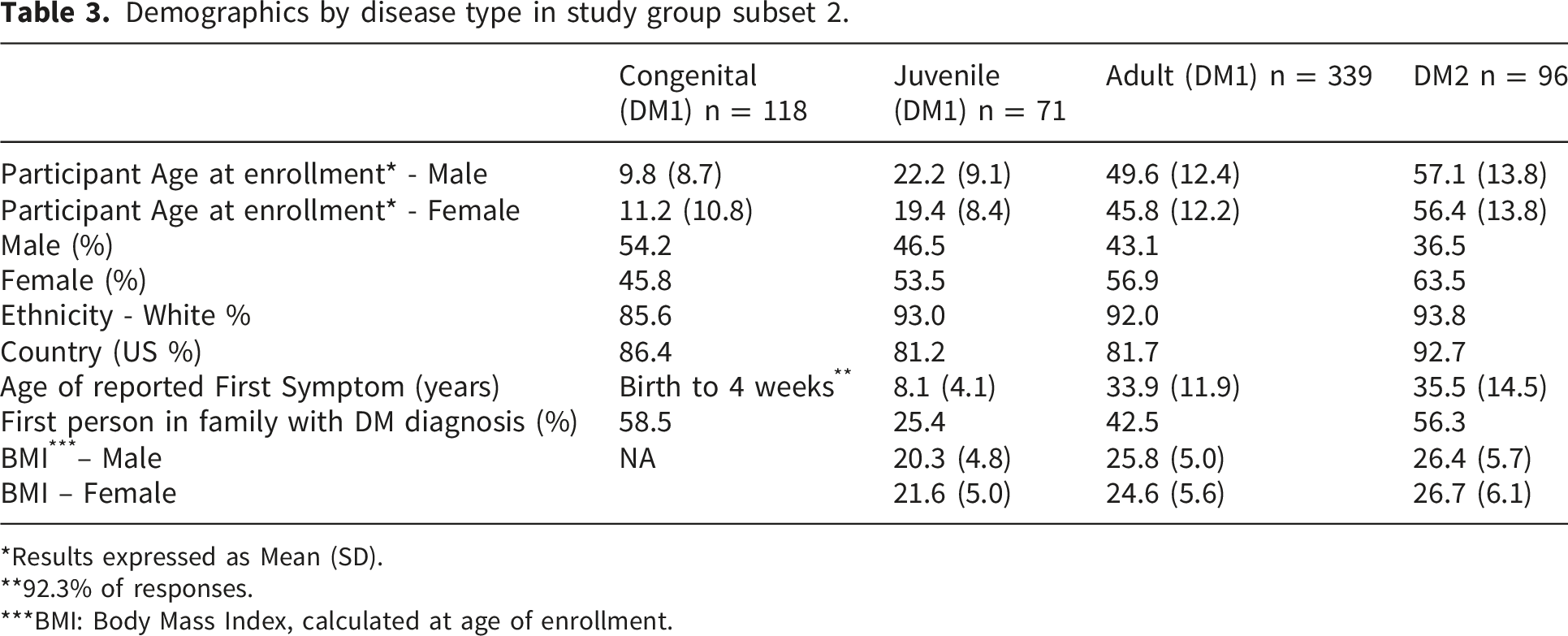
*Results expressed as Mean (SD).
**92.3% of responses.
***BMI: Body Mass Index, calculated at age of enrollment.
Overall, more than 90% of Registry participants self-identified as White, with smaller proportions identifying as Hispanic/Latino (4%) or Asian (2.3%) (data not shown). The majority of participants resided in the United States, followed by Canada (5%) and the United Kingdom (3%) (data not shown).
Family history patterns varied by DM subtype. More than half of participants with congenital DM1 (CDM) and DM2 were the first in their families to receive a DM diagnosis, whereas this proportion was lower among individuals with juvenile- and adult-onset DM1. Across all subgroups, mean body mass index (BMI) values were within the normal range (18.5–24.9).
Symptom prevalence and patient-reported impact
Daytime sleepiness was the most commonly reported symptom across all participants and DM subtypes, reported by 76.6% of individuals with DM1 and DM2 (Figure 1). Other frequently reported symptoms included difficulty swallowing (52.4%) and difficulty walking (42.3%), followed by cardiac manifestations (34.0%) and vision problems due to cataracts (32.8%). Myotonia (25.2%) and breathing difficulties (6.4%) were also reported. Notably, approximately 32% of participants indicated that fatigue had a severe negative impact on daily activities. Symptom prevalence by disease type.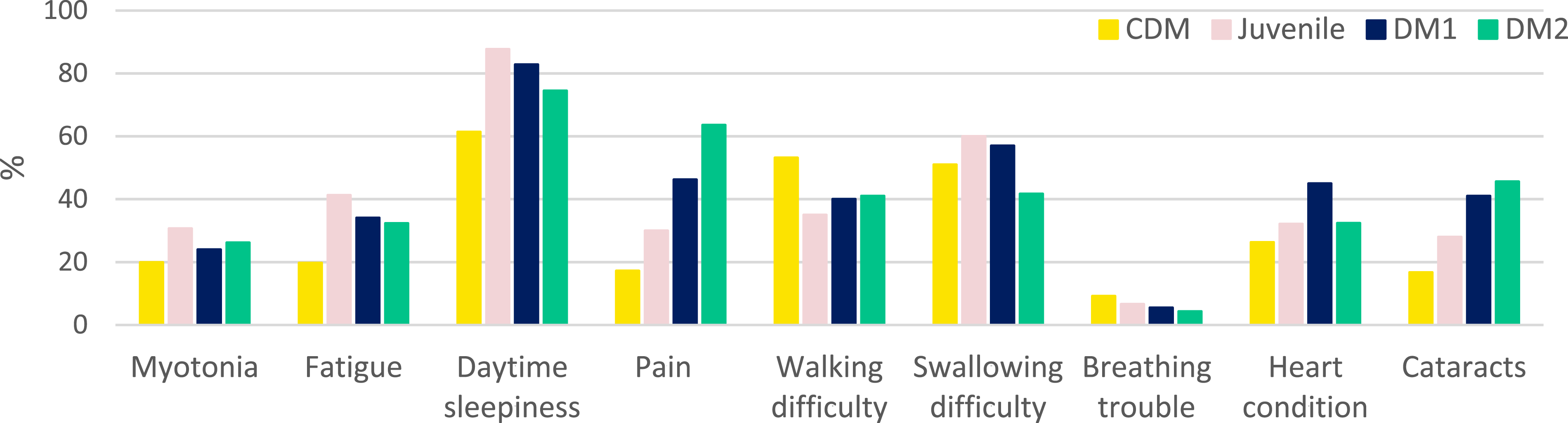
Subtype-specific analyses highlighted differences in the perceived impact of pain on quality of life. Among respondents, 63.6% of individuals with DM2 reported pain as a major burden affecting enjoyment of life, compared with 46.2% of those with adult-onset DM1.
Association between age and symptom manifestation
Relationship between age and symptom occurrence in DM1 and DM2 adult patients in study group Subset 2.
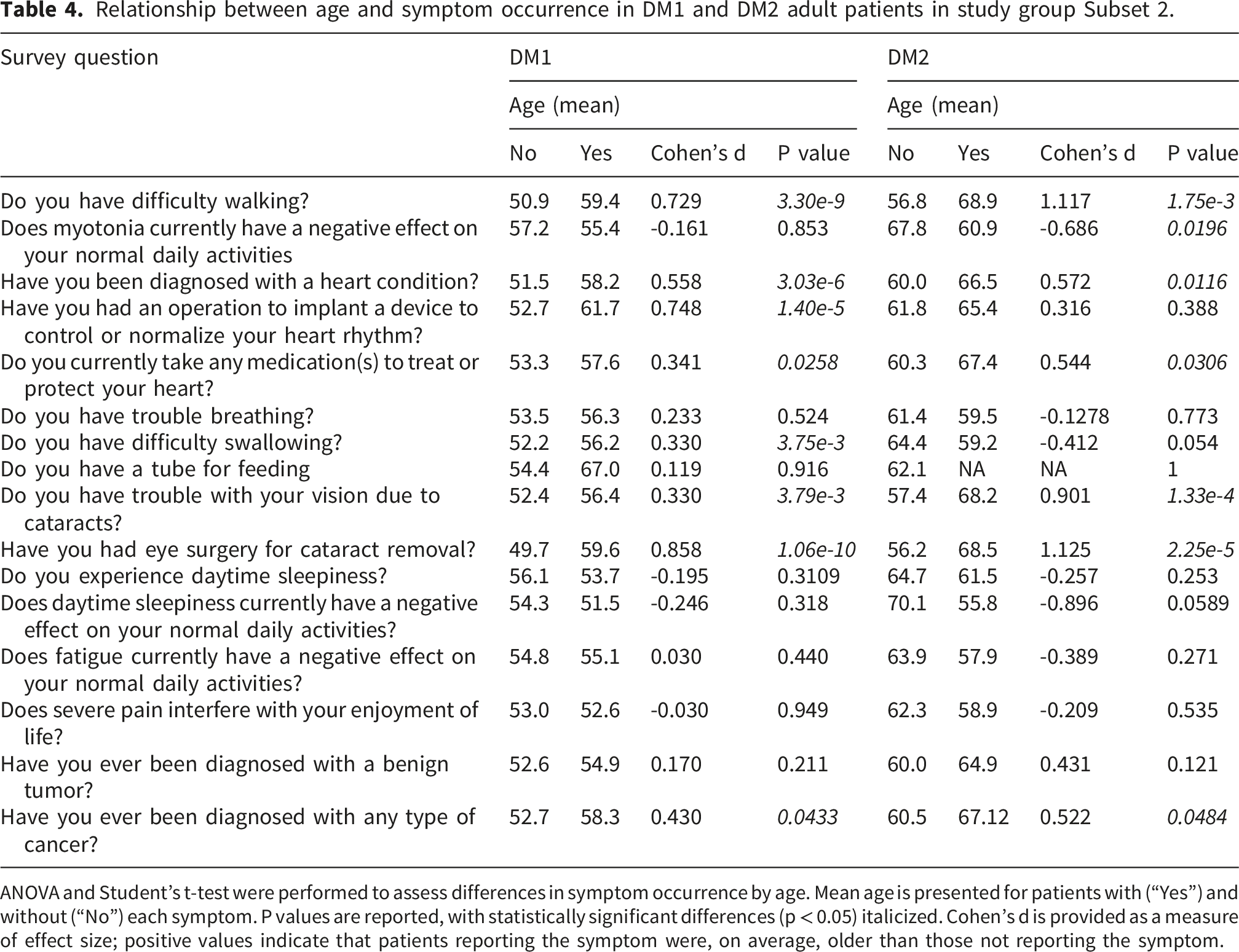
ANOVA and Student’s t-test were performed to assess differences in symptom occurrence by age. Mean age is presented for patients with (“Yes”) and without (“No”) each symptom. P values are reported, with statistically significant differences (p < 0.05) italicized. Cohen’s d is provided as a measure of effect size; positive values indicate that patients reporting the symptom were, on average, older than those not reporting the symptom.
In DM1, but not in DM2, difficulty swallowing and cardiac device implantation were also significantly associated with age. In contrast, among individuals with DM2, the reported impact of myotonia on daily activities was lower in older participants compared with younger participants.
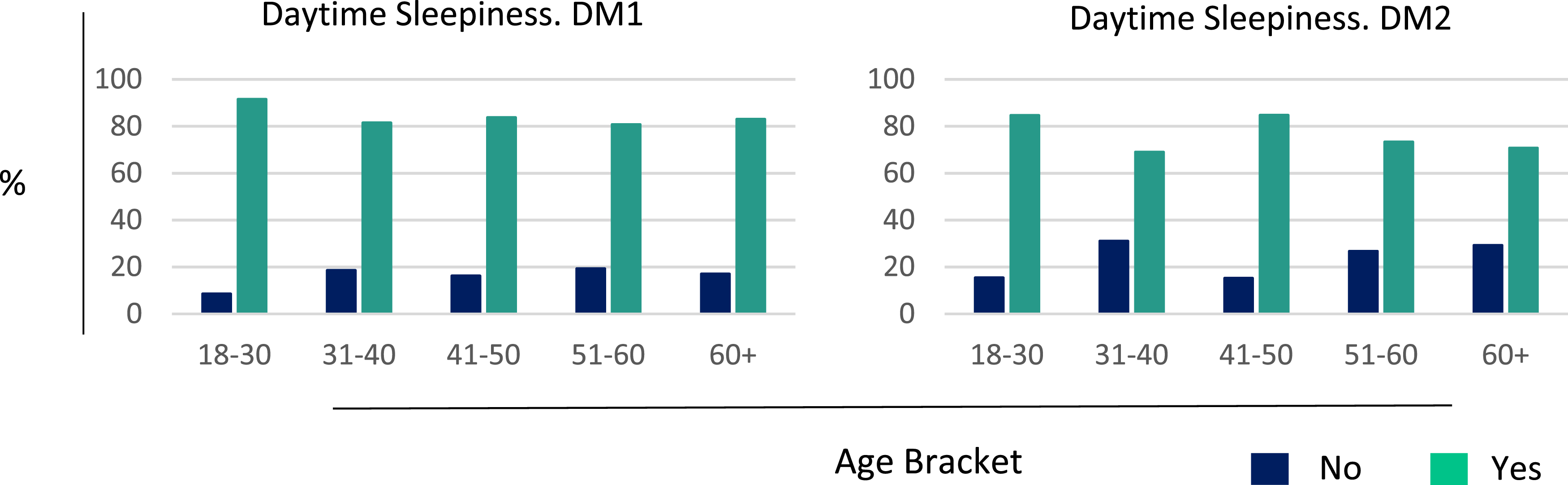
Across both DM subtypes, the prevalence of difficulty walking, cardiac complications, cataracts, and cataract surgery increased progressively with age (Figure 2; Supplementary Table 2). Daytime sleepiness was both, the most commonly reported as well as one of the earliest manifestations, affecting approximately 90% of participants and remaining prevalent across all age groups (Figure 3; Supplementary Table 2). Symptom manifestation as a function of age. Daytime Sleepiness as a function of age.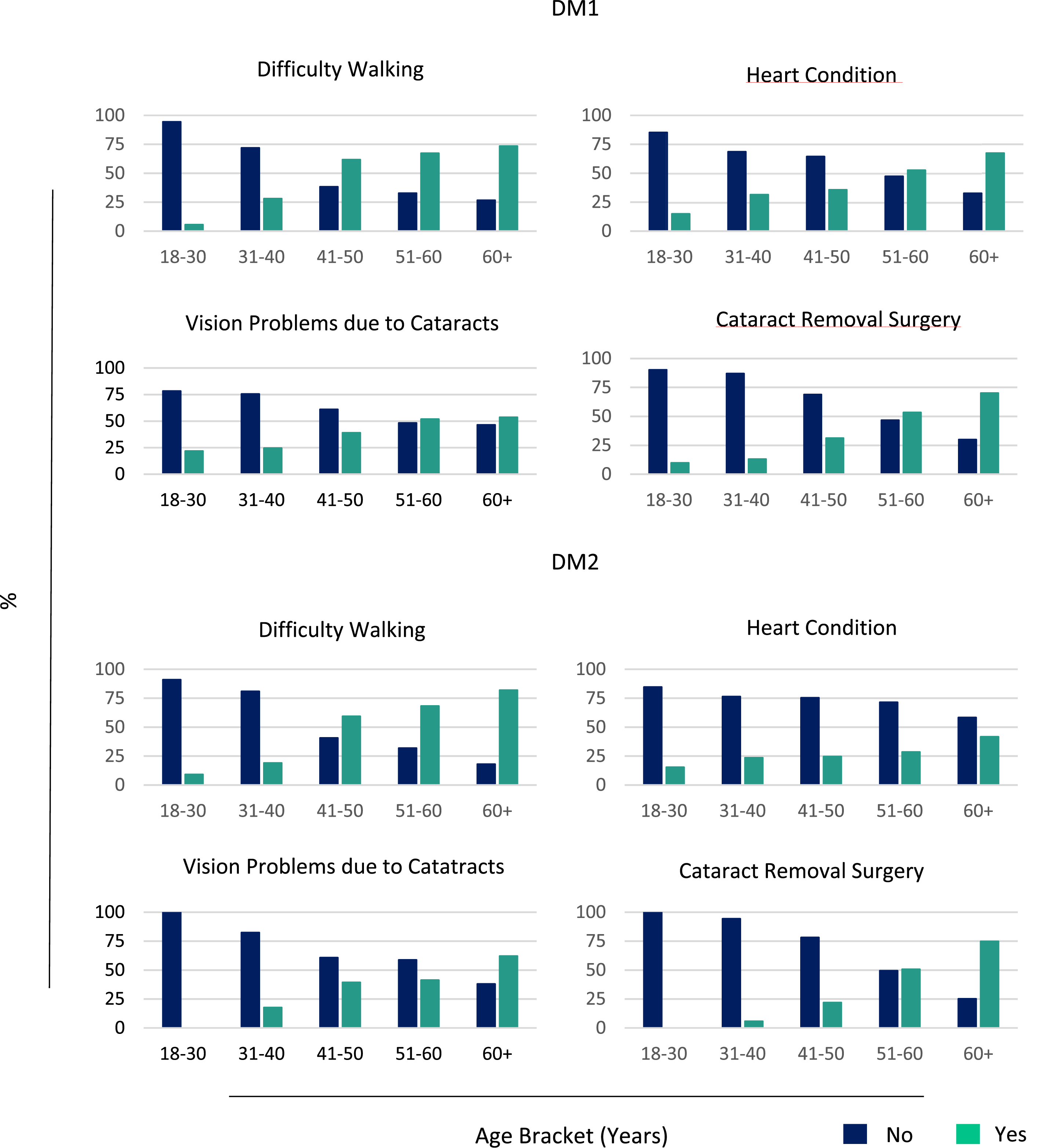
Association between repeat length and symptom manifestation
Relationship between repeat length and symptom occurrence in adult DM1 patients in study group Subset 2.
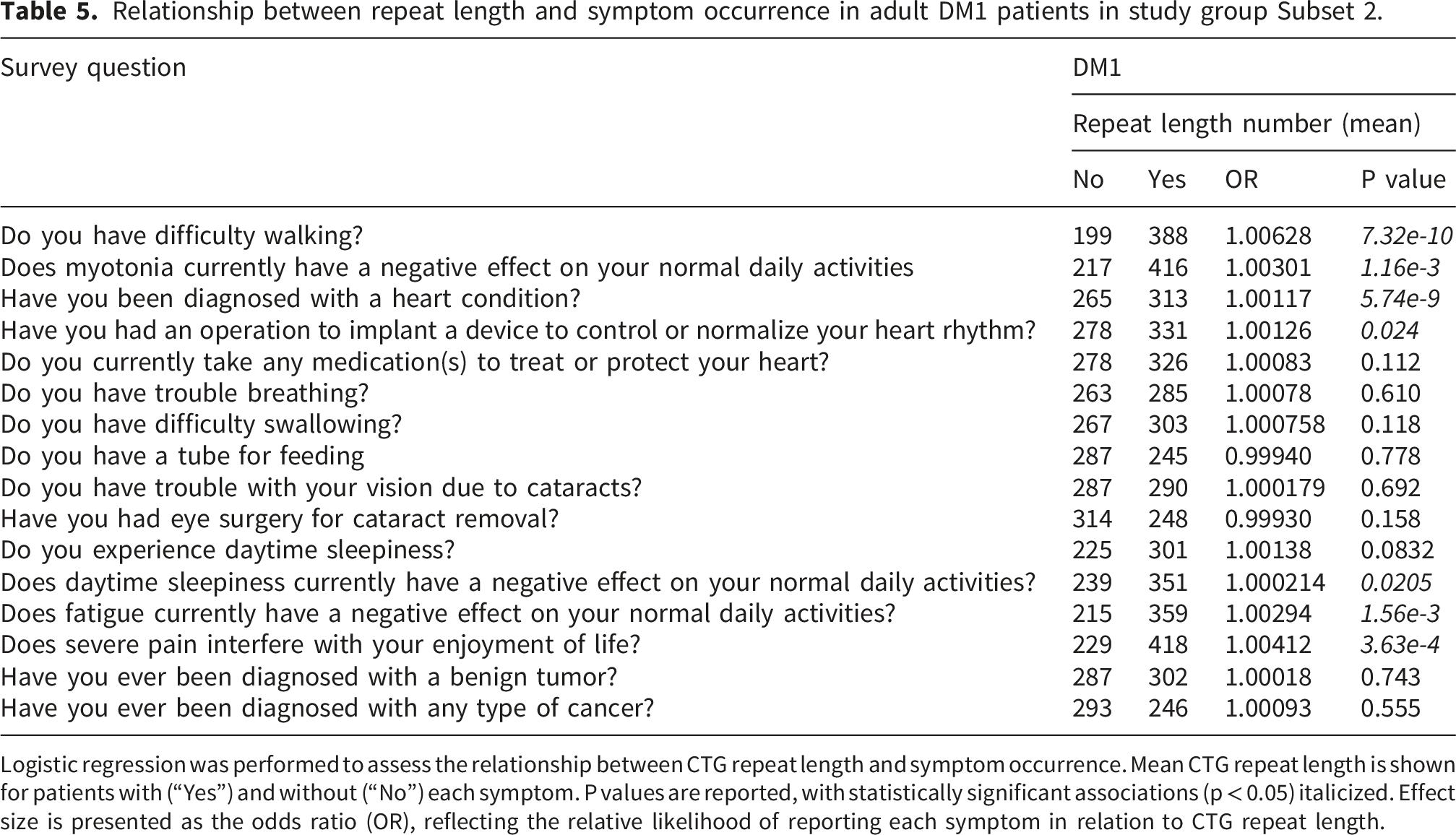
Logistic regression was performed to assess the relationship between CTG repeat length and symptom occurrence. Mean CTG repeat length is shown for patients with (“Yes”) and without (“No”) each symptom. P values are reported, with statistically significant associations (p < 0.05) italicized. Effect size is presented as the odds ratio (OR), reflecting the relative likelihood of reporting each symptom in relation to CTG repeat length.
While all Subset 2 participants had a confirmed genetic diagnosis, statistical analyses examining the relationship between CCTG repeat length and phenotype could not be performed in the DM2 cohort.
Discussion
Myotonic dystrophy (DM1 and DM2) comprises multisystem, clinically heterogeneous disorders with variability in symptom type, severity, and age of onset. This heterogeneity complicates diagnosis and challenges clinical trial design, including endpoint selection, patient stratification, and interpretation of outcomes.4,5 Improved characterization of patient-reported disease burden across subgroups is therefore essential to complement clinically derived datasets and inform therapeutic development within real-world contexts.
The Myotonic Dystrophy Family Registry provides a distinct patient-reported perspective compared with clinician-driven registries such as DM-Scope 13 and the National Registry (University of Rochester), 14 which integrate patient-reported and clinically validated data using standardized assessments. In contrast, END-DM1 represents a deeply phenotyped natural history study conducted under controlled research conditions.15,16 These complementary resources reflect trade-offs between breadth (MDFR) and clinical depth (DM-Scope, National Registry, END-DM1), underscoring the value of future harmonization and integration across platforms. The MDFR provides a large, primarily U.S.-based registry of individuals living with myotonic dystrophy. By focusing on patient-reported data, the registry characterizes the lived experience of DM, including symptom burden, diagnostic experience, age at symptom onset, and limited genotype–phenotype relationships.
The current findings complement existing clinic-based and research cohorts by validating known features of DM in a broad community-based population and by highlighting patient-prioritized manifestations, including tiredness/fatigue across DM subtypes, pain burden in DM2, and symptom onset patterns in DM1 and DM2.
Overall, the MDFR adds value by confirming and extending existing knowledge in a large patient-powered dataset and by providing a resource to inform unmet needs, clinical research, and trial readiness. Furthermore, the MDFR is extensively used as a tool supporting awareness of and recruitment for DM research studies and DM clinical trials.
Specifically, our analysis highlights several features of reported disease burden and diagnostic experience within this registry population. A substantial proportion of participants (46%) reported being the first diagnosed individual in their family, particularly among congenital DM1 (CDM) and DM2. While genetic anticipation may contribute in DM1, alternative explanations should be considered, including increased diagnostic awareness, broader availability of genetic testing, and the relatively recent establishment of the registry. In DM2, this observation is consistent with prior evidence of underrecognition associated with slower and generally milder disease progression. 17 Because we did not define or capture the date of clinical or genetic diagnosis, we cannot directly quantify diagnostic delays. However, participants reported the timing of first perceived symptoms as well as the timing of registry enrollment, allowing us to describe the interval between symptom recognition and research participation. The large difference between these time points — approximately 12 years for DM1 and 20 years for DM2— suggests a prolonged pathway from symptom onset to registry engagement. This interval may include diagnostic delay, but it is not equivalent to diagnostic delay per se. It may also reflect post-diagnosis factors such as referral patterns, registry awareness, access to specialty care or genetic testing, disease burden, geographic or socioeconomic barriers, and individual decisions about research participation. These findings, while consistent with prior reports, should be interpreted within the context of a predominantly US-based, self-selected cohort.
Symptom analyses reinforce the multisystem nature of DM and highlight patient-reported burden across domains, including central nervous system–related, cardiac, and other systemic manifestations. 18 Daytime sleepiness emerged as the most commonly reported and one of the earliest manifestations, persisting across age groups, while dysphagia, gait impairment, and cardiac involvement were also frequently reported. Pain was more commonly reported as a major contributor to reduced quality of life in DM2 compared with DM1, consistent with prior studies emphasizing the prominence of non-muscular symptoms in DM2. 18 While several manifestations increased with age, daytime sleepiness remained consistently prevalent across all age groups. These findings are broadly consistent with existing literature and suggest that some aspects of patient-reported burden—particularly fatigue and sleep-related symptoms—may not be fully captured by commonly used clinical outcome measures.19–21
Important gaps in domain coverage should also be considered. Gastrointestinal symptoms, including abdominal pain, constipation, and diarrhea, were not captured in the MDFR despite increasing recognition of their clinical relevance in both DM1 and DM2. More broadly, the registry’s symptom domains were based on consensus recommendations from the 2009 TREAT-NMD/Marigold workshop, 6 which, although enabling early harmonization, do not fully align with more recent standardized phenotype frameworks such as the Human Phenotype Ontology. 22 As a result, certain domains, particularly central nervous system and respiratory manifestations, may be incompletely represented, and symptom definitions may lack the granularity required for cross-study comparability. Ongoing efforts to update core data elements and improve alignment with standardized ontologies will be critical.23,24
Genotype–phenotype analyses in DM1 demonstrated associations between CTG repeat length and multiple clinical manifestations, consistent with prior evidence linking repeat expansion size to disease burden. 25 Repeat length information was self-reported by participants. Laboratory reports were only available for independent confirmation in 14% of the cohort, corresponding to 90 participants. Self-reported repeat length information was concordant with the laboratory-reported result in all verified cases. With no discrepancies observed in this subset, the approximate 95% upper confidence bound for the error rate is about 3.6%, indirectly supporting the validity of the self-reported repeat length data while acknowledging that more prominent reporting errors cannot be excluded in the unverified cases.
Also, several analyses were not feasible due to data limitations, including age-stratified analyses beyond primary models and subgroup analyses in congenital and juvenile DM1. In DM2, genotype–phenotype analyses could not be performed due to lack of standardization in CCTG repeat reporting, including differences in measurement units and threshold-based reporting practices. These constraints underscore the importance of standardized and comprehensive genetic data capture.
Several limitations of the MDFR should be considered. The dataset is primarily cross-sectional, with limited longitudinal participation and no analysis of repeated measures, restricting inference on disease progression. The cohort is predominantly US-based and self-identified as White, likely reflecting recruitment patterns as well as barriers related to language and access to digital or research resources. The registry was administered in English, which may have further influenced participation. Consequently, populations with distinct epidemiologic characteristics (e.g., French-Canadian populations in Quebec) may be underrepresented. Additional limitations include self-selection bias, potential misclassification due to reliance on self-reported data, and incomplete or inconsistent data capture.
Sustained registry participation in DM hinges not on patients’ willingness but on the delivery of clear, tangible value. Current limitations, including fragmented longitudinal data, limited representativeness, and a weak link to therapeutic development, constrain both engagement and impact. Addressing these challenges requires re-envisioning registries as patient-centered, development-aligned platforms in which patient experience and patient-reported outcomes are not ancillary, but foundational—integrated alongside high-quality genetic and longitudinal data, 26 enabling seamless data capture from clinical care and harmonization across initiatives. Standardization of outcome measures will be one critical element to ensure cross-study comparability and regulatory relevance, alongside strategies that directly link participation to trial readiness and individualized research opportunities. Sustainable governance models will be essential to ensure trust, scalability, and long-term impact.
Notwithstanding these limitations, the MDFR provides a valuable patient-centered perspective that complements clinically derived datasets. These findings advance understanding of patient-reported disease burden and highlight gaps in domains that remain underrepresented in current outcome frameworks. Future efforts should prioritize improved data standardization, expanded longitudinal data collection, and integration of patient-reported, clinical,27,28 and genetic datasets across platforms. As therapeutic development in DM accelerates, such integrated approaches will be essential to support patient stratification, optimize study design, and enable evaluation of clinically meaningful outcomes.
Supplemental material
Supplemental material - Myotonic dystrophy family registry. The patient experience
Supplemental material for Myotonic dystrophy family registry. The patient experience by Sofia Olmos, Danny Kuei, Kleed Cumming, Tanya Stevenson, and Andreas Rohrwasser in Journal of Neuromuscular Diseases.
Supplemental material
Supplemental material - Myotonic dystrophy family registry. The patient experience
Supplemental material for Myotonic dystrophy family registry. The patient experience by Sofia Olmos, Danny Kuei, Kleed Cumming, Tanya Stevenson, and Andreas Rohrwasser in Journal of Neuromuscular Diseases.
Supplemental material
Supplemental material - Myotonic dystrophy family registry. The patient experience
Supplemental material for Myotonic dystrophy family registry. The patient experience by Sofia Olmos, Danny Kuei, Kleed Cumming, Tanya Stevenson, and Andreas Rohrwasser in Journal of Neuromuscular Diseases.
Supplemental material
Supplemental material - Myotonic dystrophy family registry. The patient experience
Supplemental material for Myotonic dystrophy family registry. The patient experience by Sofia Olmos, Danny Kuei, Kleed Cumming, Tanya Stevenson, and Andreas Rohrwasser in Journal of Neuromuscular Diseases.
Footnotes
Acknowledgments
The authors thank all Registry participants for their support over the years, reporting the relevant clinical information used in this study. The authors also thank Drs. Belen Esparis, Haley Martinelli and Nadine Ann Skinner for reviewing this manuscript and providing constructive feedback. Special thanks to Mindy C. Kim for her assistance and engagement with MDFR members.
Ethical considerations
The MDFR has been formally approved in February 2013 and is monitored by the Institutional Review Board (IRB) Advarra. MDFR enrollees gave written consent for participation and de-identified data publication.
Author contributions
Sofia Olmos: Project administration. Data curation/formal analysis. Original draft. Writing - review & editing. Danny Kuei: Data curation/formal analysis/statistics. Kleed Cumming: Project administration. Visualization. Tanya Stevenson: Conceptualization. Supervision. Project administration. Andreas Rohrwasser: Conceptualization. Original draft. Writing - review & editing. Formal analysis. Supervision.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on request.
Artificial intelligence
The authors declare that no generative artificial intelligence (AI) tools were used in the preparation of this manuscript. All writing, editing, analysis, and figures were produced solely by the authors, without AI assistance.
Supplemental material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.