
Abstract
Stability assessment is a crucial prerequisite in pharmaceutical development, ensuring safety, efficacy, and desired control of product performance throughout its shelf life. Although conventional stability testing guided by International Council for Harmonization Q1A(R2)–Q1E and Q5C remains essential for regulatory compliance, it is often time-consuming and resource-intensive. Developments in analytical sciences and computational modeling have transformed stability programs from trial-driven processes to predictive and risk-based strategies. Novel predictive tools, such as accelerated predictive stability studies, enable rapid, accelerated, science-driven prediction of product behavior under real-world conditions. As the industry increasingly adopts biologics, biosimilars, and nano-enabled drug delivery systems, stability testing must evolve beyond conventional assays to incorporate multi-parametric, high-resolution, and functional performance-based evaluations. These tools can facilitate faster decision-making and more efficient regulatory submission, reducing delays in product availability. This review provides a comprehensive overview of advanced methodologies, novel stability-indicating methods, and the existing regulatory frameworks for novel drug delivery systems. Ultimately, the future of stability assessment lies in intelligent, predictive, and adaptive approaches that integrate advanced analytics, modeling, and robust quality frameworks.
Keywords
Introduction
The stability evaluation of a pharmaceutical drug or product is one of the most essential processes in ensuring product quality, safety, and performance throughout its lifecycle. It serves as a cornerstone in pharmaceutical development, confirming that the product remains safe and therapeutically effective during its entire shelf life.1–3 To maintain uniformity across the industry, international regulatory bodies have developed well-defined frameworks that guide how stability studies are designed, performed, and interpreted. These global guidelines specify the parameters to be tested, the duration of studies, stability-indicating properties capable of detecting changes in product quality attributes during storage, and the criteria for data evaluation for both new drug substances and finished formulations. Collectively, they provide scientifically sound evidence to justify the proposed shelf life of a product.4,5
Pharmaceutical stability refers to the ability of a dosage form to retain its quality attributes and potency until the end of its intended shelf life. Instability can arise through various mechanisms—physical, chemical, microbiological, or even therapeutic in nature. A range of intrinsic and extrinsic factors, such as temperature, humidity, pH, oxygen exposure, light, and excipient interactions (Figure 1), can accelerate degradation, causing reduced efficacy or the formation of harmful byproducts.6,7 Stability studies conducted under controlled environmental conditions—based on pharmacopoeial standards, regulatory guidelines, and industry protocols—are therefore crucial. These studies help determine the degradation kinetics of a product, guiding the establishment of suitable storage conditions, retest periods, and expiry dates. The expiry date printed on product labels is derived from validated stability data, reviewed and approved by regulatory authorities to confirm that the product maintains its safety, potency, and quality throughout its labeled period of use.3,8,9
Factors Affecting Drug or Product Stability.
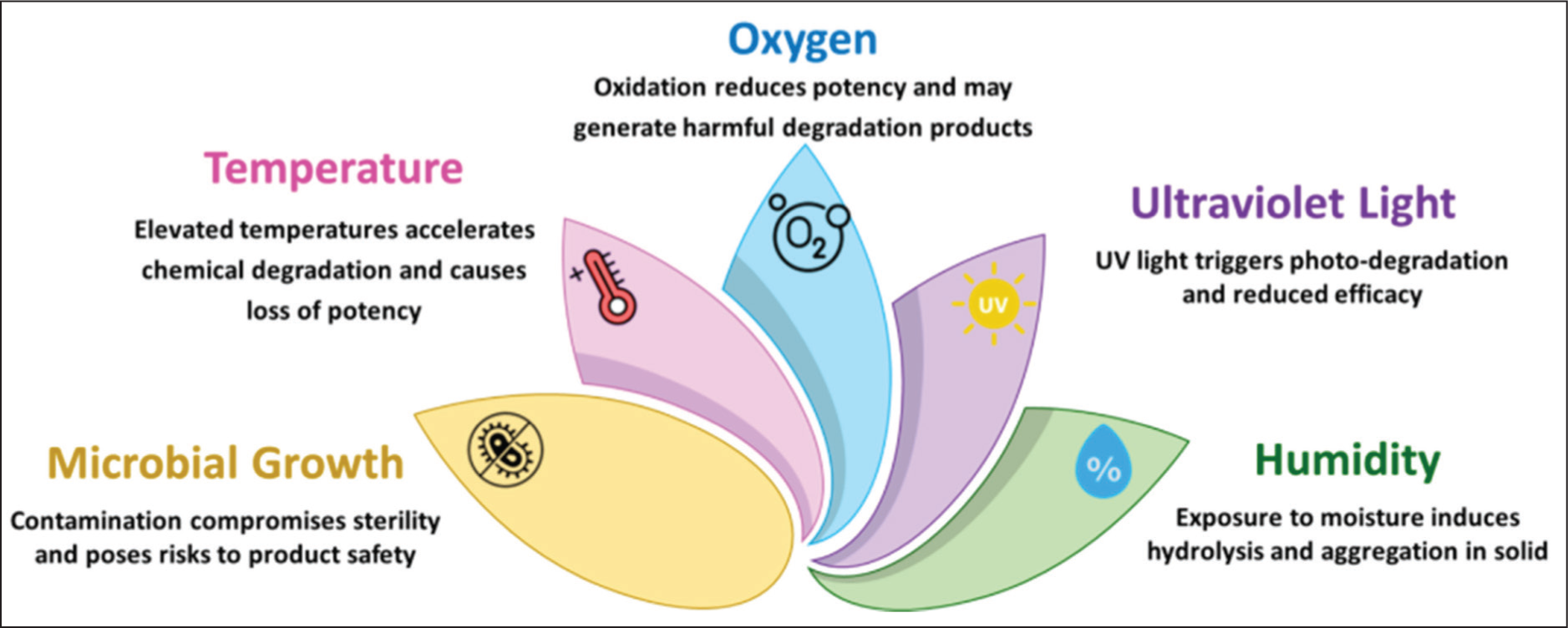
Pharmaceutical manufacturers typically implement standard operating procedures (SOPs) aligned with global regulatory expectations to ensure consistency in stability testing. However, the structure and execution of these programs can vary between organizations, depending on available resources, infrastructure, and scientific expertise. 10
Stability testing is no longer limited to conventional pharmaceuticals—it has become equally important in the cosmetic and phytomedicine industries. Regulatory agencies worldwide have issued detailed guidelines requiring stability data for herbal products as part of the registration dossier to support marketing authorization.11,12 For cosmetics, stability studies assess visual appearance, color, fragrance, pH, viscosity, and microbial safety, ensuring these products remain safe and appealing throughout their shelf life. The European Union’s Cosmetic Products Regulation (EC No. 1223/2009) specifically mandates such evaluations to confirm product quality under normal storage and usage conditions.13–15
Technological progress has significantly modernized the field of stability testing. What was once a trial-and-error process has evolved into a predictive, data-driven approach. Advanced analytical tools such as infrared (IR) spectroscopy, high-performance liquid chromatography (HPLC), and mass spectrometry (MS) now enable scientists to detect and quantify minute levels of degradation products with exceptional accuracy. 16 Additionally, computational modeling and chemometric approaches have enhanced the ability to forecast degradation trends and simulate long-term stability through accelerated studies, saving both time and resources.17,18
In the early stages of drug development, particularly during clinical trials, regulatory frameworks emphasize the need for stability data to ensure investigational products remain safe and effective throughout testing.19,20 Forced degradation and stress studies conducted during preformulation help identify stable molecules, refine formulations, and assess packaging compatibility. These preliminary results are later confirmed through formal stability studies that define the product’s labeled shelf life and facilitate global regulatory submissions. A well-structured stability testing program is essential not only for risk mitigation but also for data-driven decision-making and compliance with good manufacturing practice (GMP) standards. 17
The International Council for Harmonization (ICH) provides an internationally recognized framework, Q1A (R2)–Q1E and Q5C, for evaluating pharmaceutical stability (Figure 2). ICH Q1A outlines the general requirements for new substances and products, emphasizing both real-time and accelerated stability testing across defined climatic zones. Meanwhile, Q1B focuses on photostability studies, examining degradation under light exposure to guide appropriate labeling and packaging decisions. Products are tested under light sources that mimic natural daylight to evaluate their sensitivity to photodegradation.21,22 The current stability testing protocols focus on photostability testing during manufacturing and storage, but fail to address issues related to the photosensitivity of the drug during patient use. To overcome these limitations, several advanced approaches have been explored in recent years. These include accelerated predictive stability (APS) studies that integrate stress conditions like light, temperature, and humidity, along with kinetic modeling to predict degradation in real-world exposure conditions. Additionally, photostability tests that simulate real-life conditions, such as drug stability in the presence of moisture or post-application, have also been implemented. Collectively, all advanced methodologies provide a holistic understanding of drug behavior during light exposure, thus bridging the gap between traditional photostability testing and real-world scenarios. 23 This review aims to provide a comprehensive overview of methodological advancements in pharmaceutical stability assessment with particular focus on predictive and risk-based approaches such as APS studies. It critically examines the limitations of conventional ICH-guided stability studies and highlights the applications of advanced approaches such as the accelerated stability assessment program (ASAP), the accelerated stability modeling (ASM), and the advanced kinetic modeling (AKM). Furthermore, the review discusses the evolving regulatory landscape and the need for integration of modern tools into stability programs to enable efficient and scientific pharmaceutical development.
ICH Quality Guidelines.
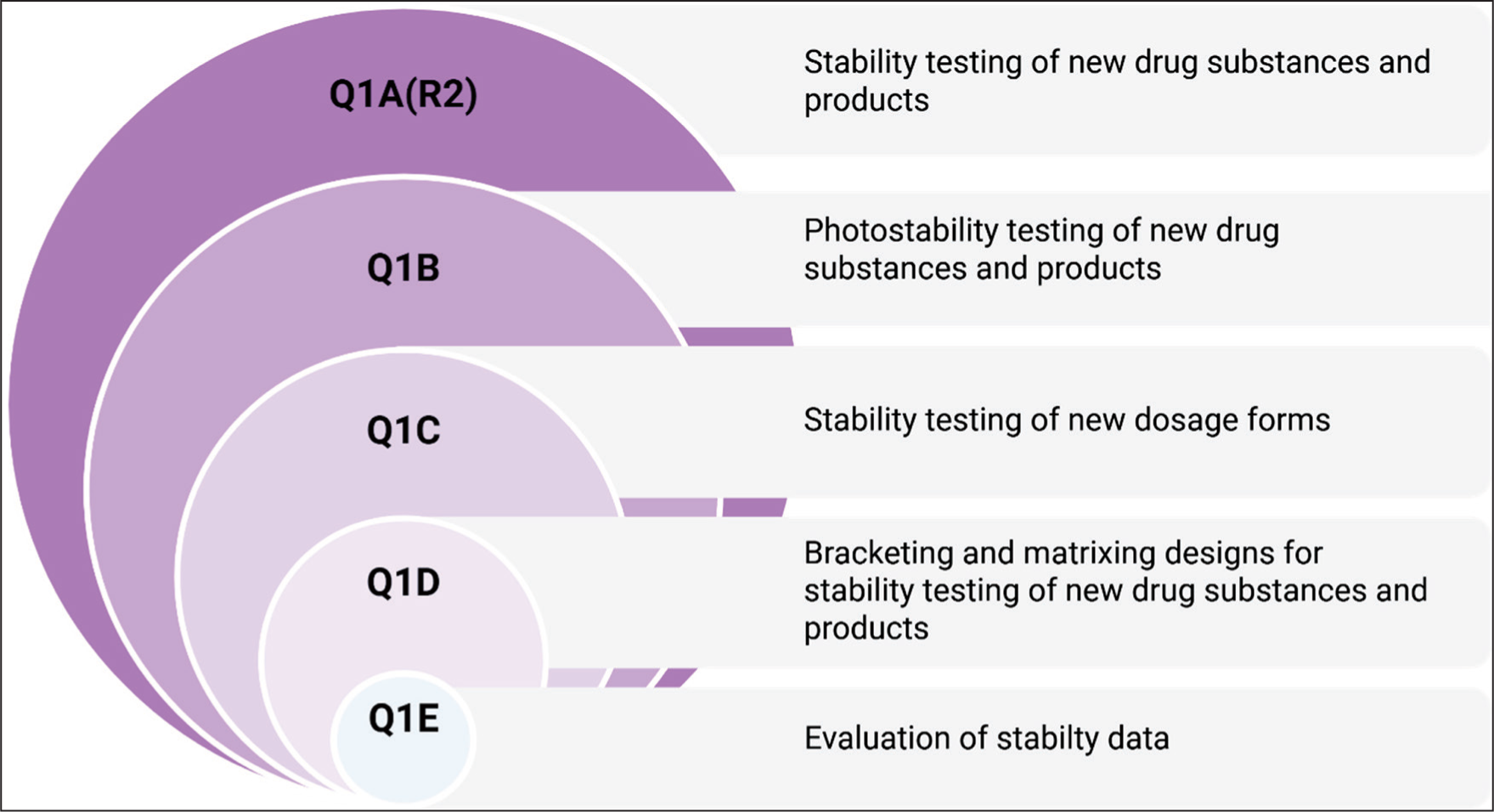
Conventional Stability Testing Methods
A combination of one or more methods aids in detecting the stability of the active pharmaceutical ingredient (API) or formulations owing to their varied physicochemical properties. These characteristics offer valuable insights for selecting suitable indicators to confirm instability and optimize parameters to ease analytical detection. Several methods are typically used to detect and study the degradation patterns of drug substances, including HPLC, ultra-performance liquid chromatography (UPLC), ultraviolet–visible (UV–vis) spectroscopy, gas chromatography (GC), high-performance thin-layer chromatography (HPTLC), capillary electrophoresis (CE), and supercritical fluid chromatography (SFC).24–29 HPLC is one of the most frequently used techniques for stability testing because of its high sensitivity, accuracy, and versatility. The compounds are subjected to forced-degradation studies in the presence of acid, alkaline, heat, sunlight and oxygen. The degradation patterns are then analyzed using a sensitive HPLC method.30–32 UV–vis spectroscopy measures the absorption of UV or visible light by a drug or product. This technique is extensively used to monitor changes in the molecular structure of a compound over time. 31 Chromatographic methods enable the separation of components within a mixture, whereas spectroscopic techniques to provide specific identification data. Hyphenated techniques integrate separation–separation, separation–identification, or identification–identification approaches. These methods are particularly valuable for characterizing impurities in forced-degradation studies, especially when isolating impurities in pure form is not feasible. 24 The hyphenated techniques include reversed phase liquid chromatography with mass spectrometer (RPLC-MS), HPLC-photodiode array UV detector (HPLC-DAD), HPLC-fluorescence detector (HPLC-FL), GC–MS, liquid chromatography–MS (LC–MS), liquid chromatography–nuclear magnetic resonance (LC–NMR), and so on. Applications of the above-mentioned techniques are summarized in Table 1.
Conventional Stability Testing Methods and Their Applications.
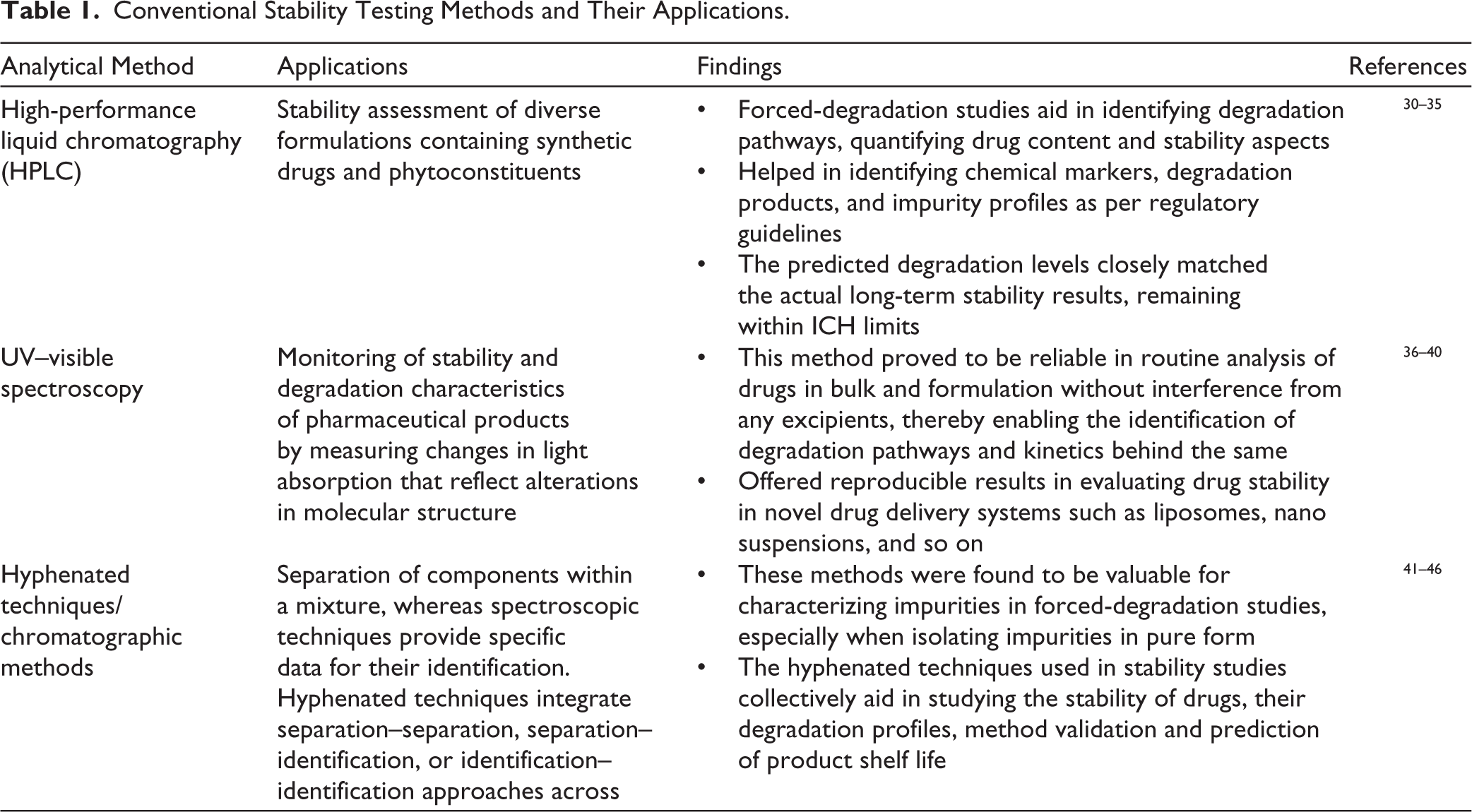
These studies demonstrate the importance of selecting the appropriate analytical techniques based on the physicochemical characteristics of the active ingredient and excipients. These conventional methods have enabled simultaneous separation, identification of impurities along with determining degradation behavior of active compounds. All these are achieved even when the actives are in trace quantities, making these traditional techniques more reliable, especially in stability assessments in pharmaceuticals.
Novel Approaches and Technologies in Stability Assessment
Due to the complex nature of biologics, novel drug delivery systems (NDDS), and so on, the conventional methods of stability assessment become time-consuming and resource-intensive. In this context, novel approaches like predictive stability modeling QbD based methods become imperative to reduce development timelines and ensure regulatory compliance.
Stability testing is imperative to assess the physicochemical parameters of a drug or product to confirm its stability, establish the shelf life of the product, and to propose suitable storage conditions. To determine these parameters, products are often exposed to various environmental conditions as specified in Figure 1 for a specific time interval. It is always monitored that the product remains stable during manufacturing, packaging, and handling. 47 Conducting stability studies is a time-consuming process that accelerates the chemical and/or physical breakdown of products that are subjected to accelerated conditions. According to ICH Q1A(R2) guidelines, long-term testing conditions should match the storage conditions proposed for the product (e.g., 25℃ ± 2℃/60% RH ± 5% RH or 30℃ ± 2℃/65% RH ± 5% RH or refrigerator at 2℃–8℃). Testing at accelerated conditions includes maintenance of the product at 40℃ ± 2℃/75% RH ± 5% RH or at 25℃ ± 2℃/60% RH ± 5% RH, for 6 months, including periodic testing at 0, 3, and 6 months for products meant to be stored at room temperature and refrigerator, respectively. Furthermore, if long-term studies are performed at 25℃ ± 2℃/60% RH ± 5% RH and a notable alteration is observed at any stage during 6 months of accelerated testing, it becomes necessary to conduct further testing under an intermediate storage condition. Products may undergo changes during transportation, which are analyzed by subjecting the product to stability studies under stressed conditions. In accordance with regulatory guidelines, the drug or product is subjected to higher temperatures for a fixed time interval (1 month) and examined for any deviations caused in comparison with the labeled storage conditions. Further real-time long-term stability studies encompassing at least 12 months in a minimum of three primary batches are performed to validate the proposed storage conditions and shelf life. 48
The ICH methodology primarily focuses on verifying a product’s stability rather than forecasting it. Consequently, it is crucial that long-term stability testing encompasses the entire proposed lifespan. 49 Traditional stability assessments are conducted according to ICH quality guidelines, which involve applying specific environmental conditions for a set duration to assess any possible physical or chemical degradation that could negatively impact product quality during storage. 50 Chemical degradation can be oxidation-mediated or due to the presence of reactive impurities such as residual peroxides, reactive oxygen species, and inorganic metals.51,52 The Arrhenius equation indicates that as the temperature rises, chemical degradation speeds up, implying that it is feasible to estimate the degradation rate at lower temperatures using data from these accelerated conditions. However, the reliability of these predictions is not guaranteed. Consequently, long-term or real-time studies are still crucial, as practical experience frequently shows variations between the results obtained from accelerated conditions and those from long-term studies. 54 However, accelerated stability studies have certain drawbacks. First, the Arrhenius equation considers only temperature, even though relative humidity (RH) is a vital factor for solid-state drug products. Additionally, for some drug products, the presence of oxygen may also play a significant role. Second, in the realm of solid-state pharmaceuticals, the API can be found in a variety of micro-environments. It may appear on the surface as a crystalline structure, in an amorphous state, or dissolved within other substances. Each of these forms exhibits unique kinetic properties, leading to an overall complex kinetic behavior. These factors may not be relevant to solutions, in which all molecules share the same microenvironment and exhibit uniform reactivity. However, changes in pH owing to temperature fluctuations can introduce complex kinetics, rendering predictions based on high-temperature data unreliable.53,54
In the product development cycle, carrying out long-term stability protocols requires substantial investments in time and resources, resulting in considerable costs. Health authorities should therefore explore and implement alternative approaches to support stability studies. Recently, various scientific and risk-based strategies have been developed, particularly for solid drug products.55–58 In 2015, the International Consortium for Innovation and Quality in Pharmaceutical Development established a task force dedicated to enhancing pharmaceutical development through the application of risk-based predictive stability (RBPS) tools. In response to the ICH Quality Discussion Group’s (QDG) suggestions to update ICH stability guidelines, the task force reviewed ICH Q1A, Q1B, Q1C, Q1D, Q1E, and Q5C. The group identified that the current guidelines fall short of effectively incorporating modern tools and strategies, such as stability risk assessment (SRA) and RBPS tools, into the development of stability programs and compilation of stability data packages for registration submissions.
These tools are used routinely in formulation development to assess stability characteristics, particularly degradation mechanisms. Stability information can be generated within weeks, which would otherwise require months or years to discern under long-term stability testing. Additionally, the data obtained from RBPS or SRA tools could be used by industries to support regulatory submissions. 59
Stress studies performed during registration stability studies often produce limited data, as they usually involve testing at only one elevated temperature for a month. This single stress condition may not cover all potential degradation pathways and conditions that could affect product stability throughout its shelf life. These deficiencies in guidance and restricted data can result in delays in the registration process, thereby delaying patient access to vital medications. 60 The group suggested conducting additional stress studies to thoroughly comprehend the effects of various stress conditions throughout the product’s shelf life. These studies should extend beyond the registration stability studies outlined in the current guidelines. These studies may have involved different temperatures and time intervals. A more comprehensive assessment of the product’s stability profile and potential degradation pathways can be achieved by broadening the scope of stress studies to encompass a wider array of conditions and time frames.
APS studies are a new method that has become more effective and time-efficient for predicting the long-term stability of pharmaceutical products. These studies are carried out to measure the changes that products undergo in storage conditions that accelerate the rate of degradation.59,61 Unlike the ICH method, APS focuses on rapidity and flexibility (accelerated), as well as scientific and risk-based forecasting (predictive). APS is characterized by diverse storage conditions, which encompass high temperatures and a wide range of RH levels. These conditions are specifically tailored according to the physical and chemical stability of the drug to ensure that the degradation mechanism aligns with the long-term storage conditions. The storage duration is flexible, ranging from a few days to several weeks, depending on the storage conditions and the drug’s stability. The number of time points can vary from one to several, based on the intended application. The endpoints of the study are not determined by time but are instead linked to the specification limit of the shelf life-limiting attributes, such as degradation product or assay loss. As a result, the storage time should be shorter under more severe conditions and longer under milder conditions to achieve a similar level of degradation. Degradation is assessed by fitting the data to a modified Arrhenius or an empirical equation, using either a model-free approach or a specific kinetic model. Statistical analysis is performed to assess the quality of the model’s fit. Long-term storage stability is predicted for rank order, or if appropriate, for retest period or shelf life, using the APS model developed from APS storage condition data. The APS model can be validated using real-time stability (ICH) data for registration or post-approval purposes. Emerging science and risk-based stability programs, such as ASAP and ASM, are known as APS approaches. The ASAP is considered a model-free approach, whereas ASM employs specific kinetic models. These have been effectively adopted by pharmaceutical companies for various applications.62,63
Accelerated Stability Assessment Program
This program was developed by Waterman in 2007 for modeling the stability of pharmaceutical substances, using a modified Arrhenius equation and a model-free isoconversional (IC) approach. 64 It presents practical guidelines for performing routine stability assessments in a typical pharmaceutical analysis laboratory. Although real-time studies, as recommended by ICH, are frequently employed to evaluate the stability of pharmaceuticals, a faster assessment can be achieved using an accelerated method.65–69 ASAP is an expedited technique designed to determine the IC time, or the time to failure, under various temperature and humidity conditions. 70
The successful implementation of ASAP in Pfizer, along with the commercial availability of ASAPprime→, a software specifically developed to support ASAP studies (developed by FreeThink Technologies, Branford, Connecticut), has been highlighted in numerous scientific articles evaluating APS studies. 63 The primary benefit of ASAPprime is that it allows individuals, even those lacking formal statistical education, to perform ASAP stability modeling, with the process being completed in just a few days or weeks. Recognizing that molecules take longer to break down under mild conditions, the ASAP strategy was to replicate the chemical degradation of drugs by using accelerated conditions. The experimental ASAP program involved five to eight storage conditions, with temperatures set between 50°C and 80°C and RH ranging from 10% to 75%. The design was intended to speed up degradation within a shorter timeframe of 3–5 weeks by subjecting the drug product to different combinations of temperatures and humidity levels. ASAP was proposed to encompass five elements: the IC concept to tackle the intricacies of solid-state kinetics; an Arrhenius equation modified for moisture, which explicitly presumes an exponential link between RH and reaction rate; statistical analysis that estimates parameters and clearly defines error margins for predicted shelf lives; the integration of RH’s impact on stability with the protection offered by packaging to assess shelf life across different storage conditions and packaging configurations; and the ability to identify the critical quality attributes (CQAs) that influence drug stability.54,71 The use of an appropriate statistical method and the most suitable kinetics model helped to determine the shelf life of the drug product. The ASAP method was also utilized to support the selection of an appropriate primary packaging configuration. This approach was less time-intensive and required fewer resources than the conventional ICH accelerated stability method. It was used in clinical phase settings to ascertain a product’s shelf life and assess various primary packaging alternatives. Subsequently, ASAP studies have been utilized for assessing drug stability with various APIs,72,73 alterations in excipients, 74 modifications in packaging configurations, 55 and process parameters.75,76
To prevent stability issues during pharmaceutical product development, it is imperative to predict stability during the early stages of development. Table 2 highlights case studies involving the use of ASAP in the early stages of development of various dosage forms.
ASAP and Its Applications in Formulation Development.
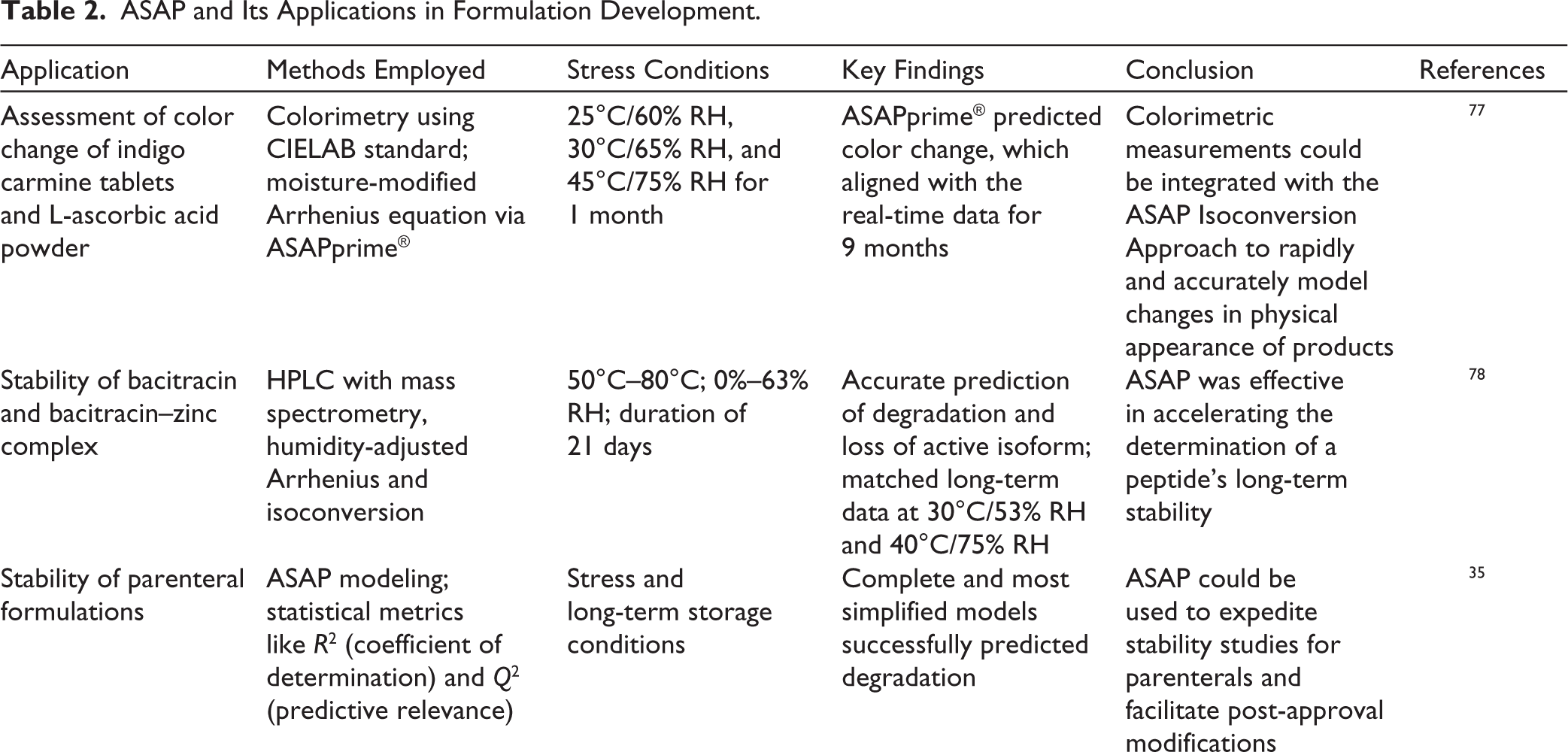
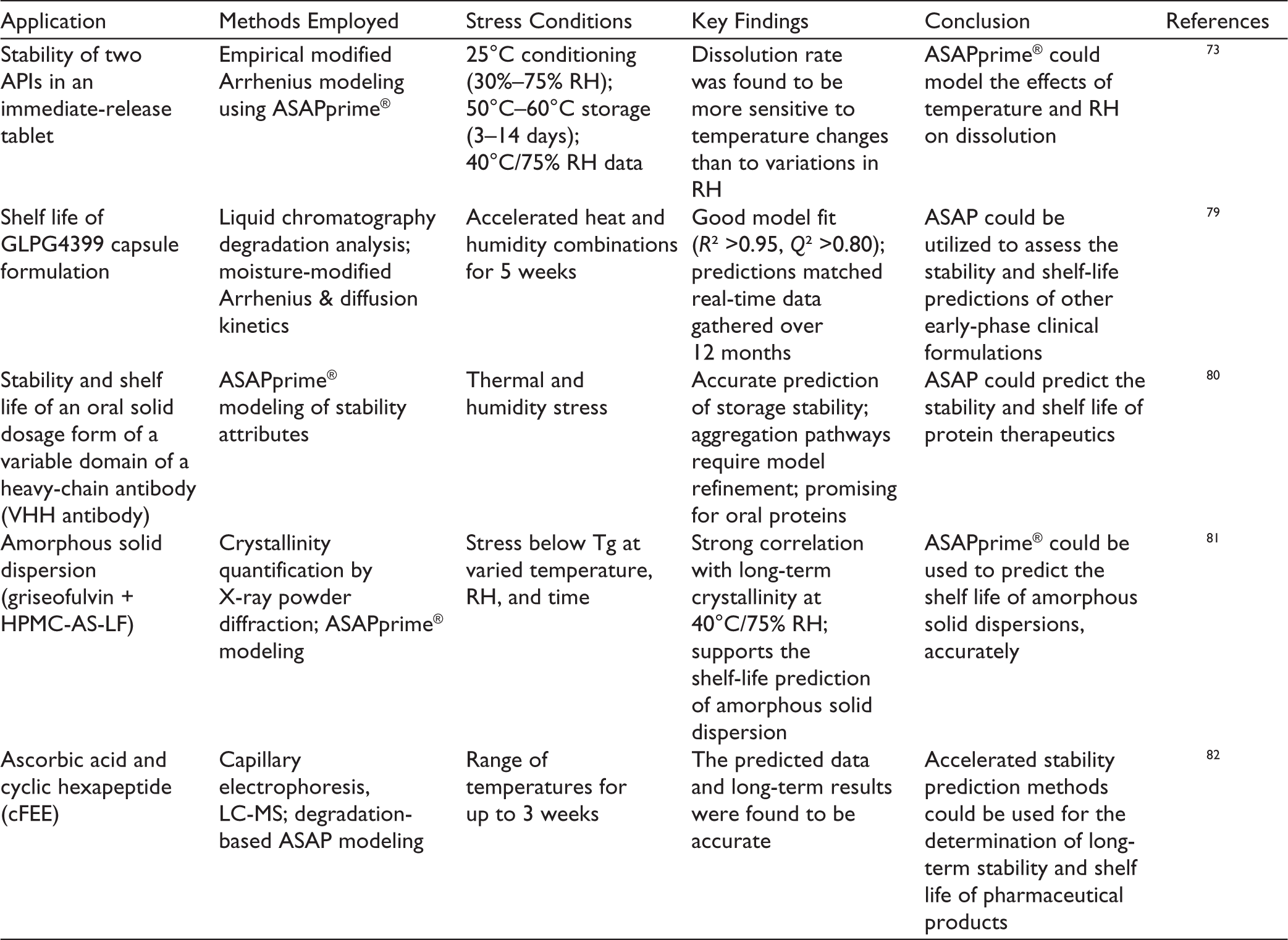
The studies collectively demonstrate that ASAP is a robust and versatile tool for predicting the long-term stability and shelf life of a wide range of pharmaceutical products, including small molecules, peptides, proteins, parenteral, and so on. By integrating stress testing with advanced analytical testing under elevated temperature and humidity conditions, ASAP enables rapid and reliable estimation of degradation pathways, physical changes and CQA. The models showed strong agreement with real-time stability data across multiple platforms, ranging from chemical degradation and dissolution behavior to physicochemical characterization. Additionally, ASAP facilitates informed decision-making in product development, formulation optimization and expedites regulatory approvals. Despite certain challenges involved with protein molecules, the overall evidence highlights ASAP as a powerful approach to reduce development timelines and accelerate stability studies.
Accelerated Stability Modeling
Combining accelerated stability testing with modeling to forecast the stability of compounds, mixtures, and products under long-term storage conditions offers substantial advantages for science-driven decision-making throughout the development of drug substances and products. The study can be completed within three working weeks, and the insights and information gained during this period can be as valuable as years of conventional analysis.
Clancy et al. reported an ASM approach (kinetic modeling concept) used in the GlaxoSmithKline R&D department. 59 Moisture vapor transmission rate models were utilized along with kinetic models to simulate various packaging types, thereby enhancing the approach’s applicability. By assessing the impact of packaging and desiccants, control strategies can be formulated. This method allows the same stability data to be applied to multiple packaging types and different “climatic zones,” which conserves resources and accelerates development time when different variations need to be established for long-term stability to generate data supporting product decisions. The authors determined that water should be considered a reactant in a kinetic model rather than an exponential parameter. This conclusion is based on the observation that products sensitive to humidity, when subjected to significant desiccation, exhibit reduced degradation rates as predicted by the concentration of water in the system raised to a power, rather than as an exponent of RH. For products that can be effectively packaged with a desiccant, as indicated by a kinetic equation, an equation assuming an exponential RH relationship would lead to more costly refrigerated storage. 62
Advanced Kinetic Modeling
The ICH guidelines recommend the use of linear or nonlinear regression and statistical modeling, to estimate shelf life. Simple kinetic models like zero or first-order mechanisms are often employed to determine product degradation rates through accelerated stability programs, which subject products to higher temperatures than usual (typically 5°C, 25°C, 37°C, or 40°C). Although these methods have been successful for small molecules and complex biologicals such as vaccines, they fail to describe the complex, multi-step reactions involved in these biologicals. 83 One way to address this issue is to employ AKM, a technique that utilizes kinetic models capable of depicting the degradation rates of products, regardless of the complexity of their degradation pathways. 84 This technique could be used to forecast the stability profile of biologicals such as vaccines, oligonucleotides, recombinant monoclonal antibodies, and in vitro diagnostic products.
AKM is a contemporary predictive approach employed in the development of biopharmaceuticals to precisely simulate and predict the stability of biologics, vaccines, and other intricate bio-products across different temperature settings. The concept of AKM is based on Arrhenius-based kinetic equations, which characterize chemical and physical degradation processes based on temperature, time, and reaction order, and encompass both single-step and multi-step processes. It utilizes data from accelerated stability studies that are usually performed at higher temperatures (e.g., 25°C, 37°C, 45°C) to accurately predict product behavior at lower, real-world storage temperatures. By aligning experimental data with one or two-step kinetic models, AKM can depict both single and multi-pathway degradation mechanisms.85–87 This method has proven to be effective in predicting long-term shelf-life from short-term data (3–6 months), optimizing stability-indicating attributes of biologics (e.g., aggregation, depolymerization, antigen loss), comparing batch stability for manufacturing consistency, supporting regulatory submissions (European Medicines Agency [EMA], US Food and Drug Administration [USFDA] acceptance in select cases), and managing temperature excursion assessments and cold-chain logistics. Pharmaceutical companies such as Novartis, Sanofi, and AbbVie have utilized AKM for both liquid and freeze-dried biotherapeutics, achieving precise long-term stability forecasts that have been confirmed with experimental data spanning up to 3 years. Regulatory authorities such as EMA and Health Canada have approved stability claims based on AKM in product submissions.85,88 Fundamentally, AKM merges the principles of chemical kinetics, thermodynamics, and statistical analysis to establish a robust and scientifically validated framework for predicting the stability of biologics. This methodology enables quick, data-driven decision-making in the areas of formulation development, shelf-life determination, and risk management throughout the entire lifecycle of the product.
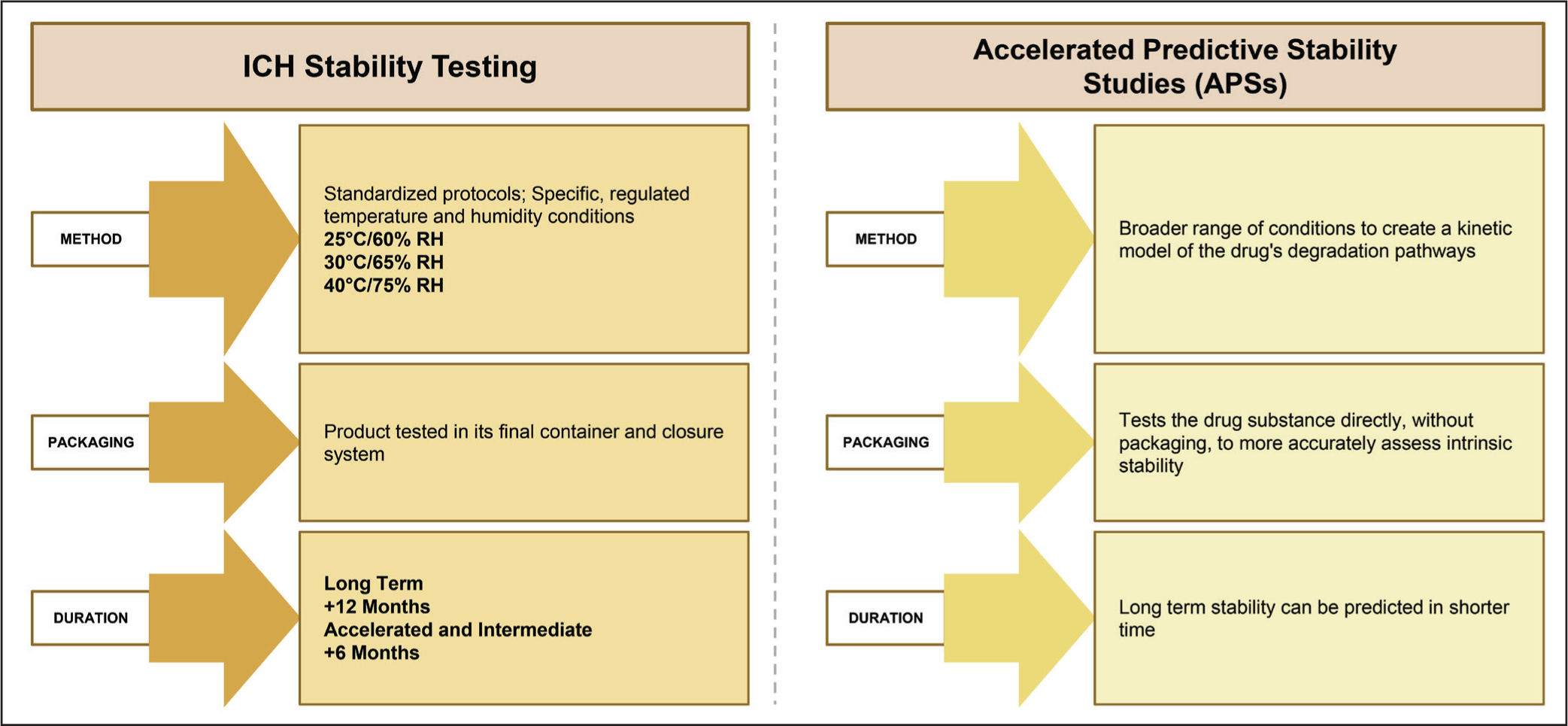
Clénet et al. applied the AKM approach to investigate the degradation kinetics of a protein-based (antigen PhtD adsorbed to phosphate-treated aluminum hydroxide) vaccine, a virus-based vaccine (ALVAC) and an emulsion-based adjuvant formulation. Accelerated stability studies were performed at temperatures of 5°C, 25°C, 37°C, and 45°C and the resulting data were analyzed using kinetic models (one-step or two-step). Statistical analysis was used to determine the most appropriate model, which was then used to predict the long-term degradation under desired storage conditions. These findings showed that vaccine stability can be determined accurately. For the protein-based vaccine, an nth order reaction model proved to be the best fit to describe the rate of degradation, as the model showed lower prediction errors in data collected at 25°C, in comparison to traditional first-order kinetic models. In contrast, degradation of the virus vaccine followed a two-step mechanism, which is consistent with the complex behavior observed in microorganisms. Degradation of the emulsion formulation was described by the autocatalytic kinetic model. Overall, the kinetic modeling approach could be practically used for shelf-life prediction, assessment of the impact of temperature deviations from the “cold chain,” comparison of manufacturing batches, and evaluation of stabilizers during formulation development. The authors recommended that further studies be carried out to explore how individual components of the vaccine contribute to and interact within the overall degradation process. 83 Figure 3 depicts a comparison between the traditional stability testing methods and APS studies.
Contrasting Traditional Stability Testing Protocols with Accelerated Predictive Stability Methodologies.
Limitations of APS Studies
APS studies have made great progress in predicting degradation models and reducing the costs of stability studies in comparison to traditional ICH studies. However, when both approaches are compared, the predictions are not always in agreement with each other in terms of expected degradation. The protocols are concerned with the study of chemical degradation of API and often overlook physical or physicochemical changes that may occur during stability testing, following the Arrhenius equation. Extreme temperatures should be used in APS studies; however, these temperatures should not exceed the melting point of the API. 59
Though the ASM model is projected as an efficient method for stability assessment, it has certain limitations, which should be considered when stability assessment is carried out at high temperatures. High-temperature stress testing may result in variation in pH, release of dissolved gases or evaporation of volatile solvents, with subsequent changes in API, resulting in inaccurate stability testing. Similarly, residual moisture present in lyophilized products may be removed when the sample is exposed to high temperatures. Factors such as RH and oxygen level, which are likely to impact stability, are not considered using the Arrhenius model when testing liquid or lyophilized reagents. Degradation factors such as light are not considered in the ASM, which is a concern for certain molecules that are susceptible to photodegradation more than thermal degradation. ASAP is most often used for testing chemical degradation and generates inconsistent results when used to test physical changes (such as melting, conversion from hydrate to anhydrous form, etc.). Furthermore, ASAP cannot be applied to large molecules such as proteins, as not all changes in the molecule structure are irreversible and not all variations affect the activity of the molecule.89,90
Stability Assessment of NDDSs Biologics and Biosimilars
Stability testing verifies that a biopharmaceutical product retains its quality, safety, and efficacy throughout its intended shelf life. This is a critical process for determining how these parameters change over time when exposed to various conditions such as humidity, temperature, and light. This enables developers to propose shelf life and define appropriate storage conditions for the product. Additionally, stability studies provide crucial data for regulatory submissions, demonstrating the consistency, durability, and reliability of the product under varied environmental conditions.91,92
Biological molecules like proteins and peptides, are susceptible to degradation pathways (Figure 4) like aggregation, oxidation, and deamidation. 93 The primary focus in stability assessment of biologics is understanding the degradation mechanism. Statistical methods such as regression analysis and advanced tools like analysis of covariance (ANCOVA) help in modeling these degradation trends (Table 3). The ICH Q5C guideline provides the primary framework for stability assessment of biotechnological and biological products.93,94 Stability protocols must evaluate CQA such as potency, purity, molecular characterization, and product characteristics, for the drug substance and drug product. 95
Selected Degradation Mechanisms Relative to Product Attributes.
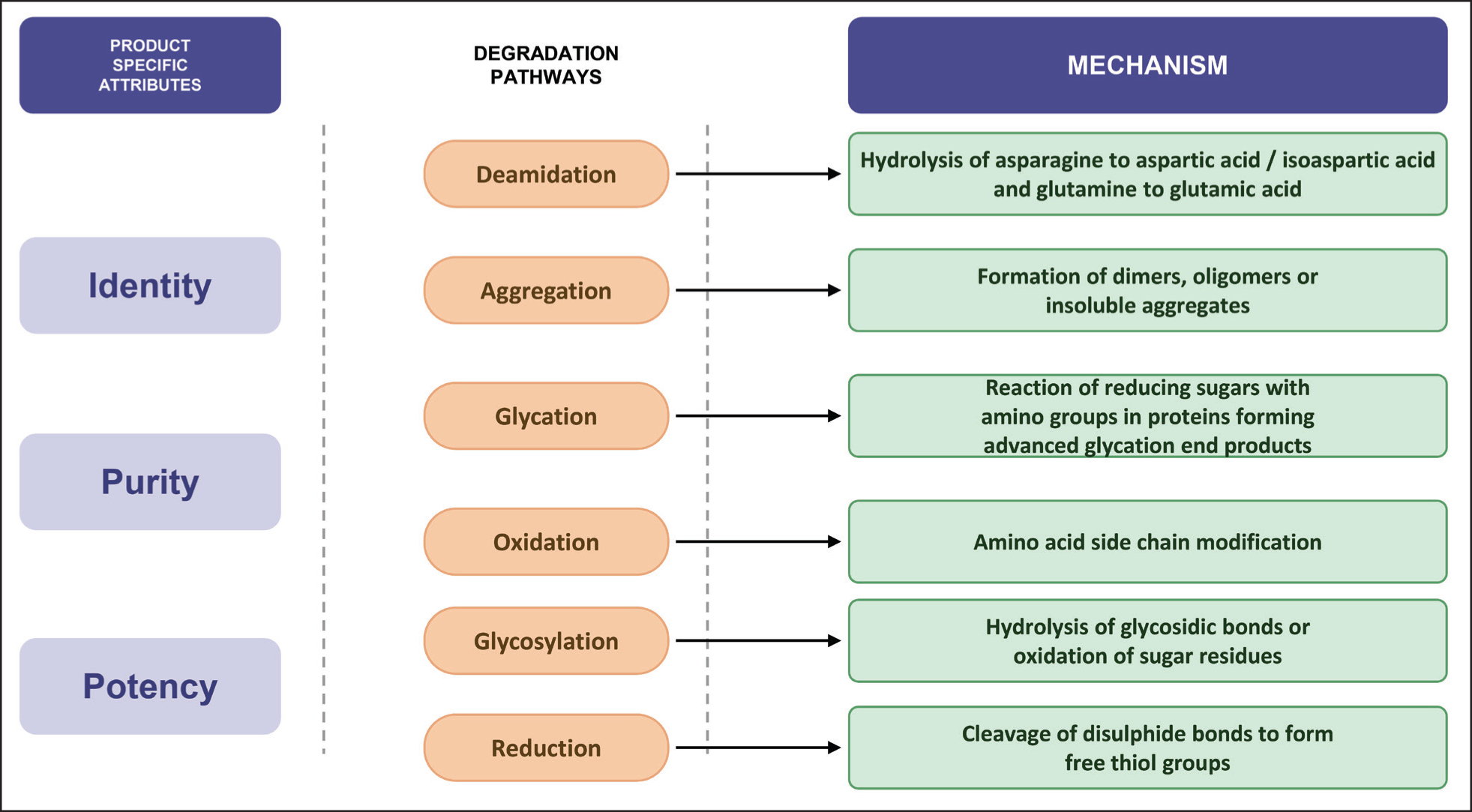
Primary Degradation Effects and Detection Techniques of Biologics.

Models such as the Arrhenius equation are valuable tools for assessing the long-term stability of biological products using data collected under accelerated storage conditions. 88 Modern stability testing methods employ state-of-the-art analytical methods, rigorous statistical analysis, and sophisticated predictive models, enabling precise determination of expiration dates.
Nanoparticles and Vesicular Drug Delivery Systems
Nanoparticle stability is determined based on the preservation of certain nanostructural characteristics such as shape, size, aggregation state, composition, surface chemistry and crystallinity. Furthermore, the optical, electric, magnetic, and biological properties of nanoparticles are dependent on these properties. Colloidal stability and aggregation affects biological properties such as toxicity, in vivo interactions, and their ultimate fate. 116 The definition of the stability of nanoparticles also varies according to their intended applications. For example, the catalytic activity of palladium nanoparticles depends on the metal surface area, suggesting that preventing aggregation of particles during shelf life is of primary concern, and hence, its stability is dependent on this. Similarly, the activity of silver nanoparticles was dependent on the retention of atomic silver in the particles.117,118
Nanoparticles possess a high surface-area-to-volume ratio and high surface energy thermodynamically unstable. The particles tend to aggregate, resulting in their nucleation and growth. The interactions between particles are governed by attractive van der Waals forces and repulsive forces. The nanoparticle dispersion is stabilized when there are adequate repulsive forces to counteract the attractive forces. Surface modification via electrostatic and steric inhibition can limit particle aggregation. 119
The size assessment of nanoparticles is one of the CQAs that defines the quality of nanomedicines. Particle size analysis of nanoparticle (NP)-based products is crucial for assessing how particle size changes over time, and providing insight into product shelf life, nanoparticle stability, and variations in drug loading. The size also influences a nanoparticle’s capacity to accumulate in specific tissues (such as tumors) through mechanisms like the enhanced permeation and retention (EPR) effect. Unlike small-molecule drugs or proteins, NP-based delivery systems cover a broad range of nanometer-scale sizes and are typically polydisperse. Moreover, these systems are usually dispersed in complex aqueous media, requiring appropriate processing to maintain colloidal stability. The presence of agglomerates must also be detected and characterized to ensure manufacturing quality control, therapeutic efficacy, and product safety. 119 The common techniques used in the stability assessment of nanoparticles120,121 are depicted in Figure 5.
Nanoparticle Characterization Techniques.
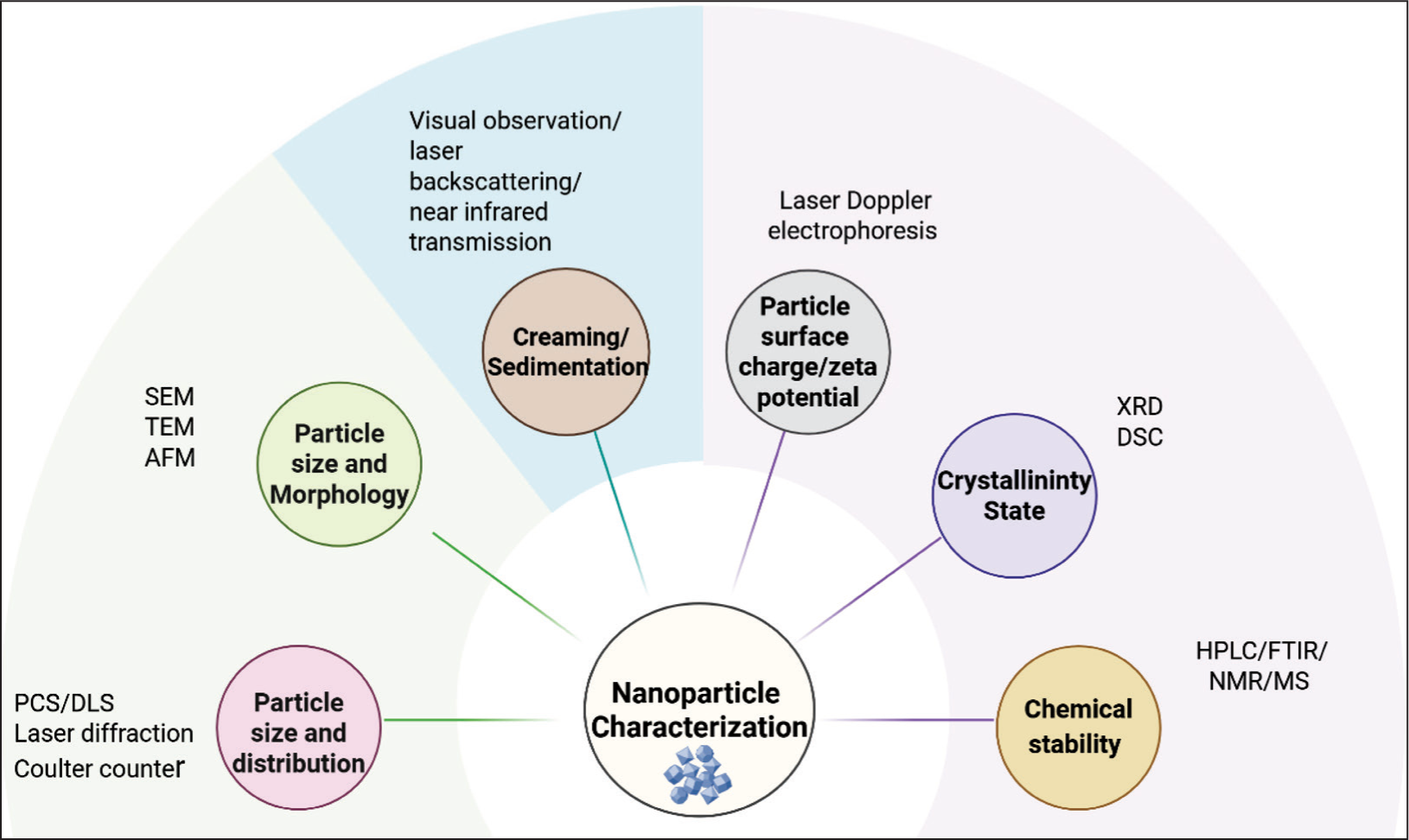
The mean particle size and size distribution of nanoparticles in liquid samples are determined by Photon Correlation Spectroscopy (PCS), also known as Dynamic Light Scattering (DLS). 122 The particle size distribution, representing the range of sizes in a dispersion, is indicated by polydispersity index (PDI). Ideally, for a narrow size distribution, the PDI value should be in the range 0.1–0.25. Values greater than 0.5 refer to a broad distribution. A limitation of this technique is that it is not applicable to solid samples. Laser Diffraction (LD) is used for both wet and dry samples at a wide detection range, ranging from 20 nm to 2000 μm. The coulter counter, in contrast, determines the exact number of particles per unit volume across various size ranges and offers greater precision than LD. SEM, TEM and AFM techniques are direct visualization techniques that can evaluate the morphology along with particle sizes. However, these techniques require additional sample preparation such as coating, which could be destructive to the particles, potentially causing changes in particle properties.123,124
Creaming and sedimentation are types of physical instabilities observed in nanoformulations such as nanoemulsions, nanodispersions, or nanosuspensions. They occur due to the gravitational separation of dispersed particles or droplets from the continuous phase over time. Creaming occurs when the dispersed phase (usually less dense particles or droplets) moves upward in the formulation due to buoyancy. Sedimentation occurs when the dispersed phase (typically denser particles) moves downward under the influence of gravity. Assessment of creaming and sedimentation in nanoformulations involves visual, analytical, and accelerated stability tests. Techniques such as centrifugation, Turbiscan analysis (Multiple Light Scattering), particle size monitoring, zeta potential, and rheology collectively provide a comprehensive understanding of formulation stability. 125
Zeta potential is a key indicator of the surface charge and colloidal stability of nanoparticles. It represents the electrical potential at the slipping plane, that is, the boundary between the stationary layer of the fluid attached to the particle surface and the surrounding bulk fluid. This helps determine the stability of nanoparticle dispersions; higher absolute zeta potential values (typically > ±30 mV) indicate strong electrostatic repulsion between particles, thus minimizing aggregation. It provides insights into the surface modification, coating efficiency, and interaction with biological membranes or other charged species. It can be used to monitor formulation consistency and predict the shelf life of nanomedicines. The zeta potential is commonly measured using electrophoretic light scattering (ELS) or laser Doppler electrophoresis. In these methods, an electric field is applied to the nanoparticle suspension, and the movement (electrophoretic mobility) of the particles is measured. The zeta potential is then calculated using the Smoluchowski or Henry equation, depending on the medium and particle size.125,126
The assessment of crystallinity in nanoformulations employs a combination of thermal, spectroscopic, and diffr-action techniques. XRD and DSC are used as primary quantitative tools, while FTIR, Raman, and solid-state NMR offer molecular-level insights, and SEM/TEM provide visual confirmation of crystal morphology. 127
The chemical stability of nanoformulations is a vital determinant of their safety, efficacy, and shelf life. It is influenced by factors such as the particle surface chemistry, excipient interactions, environmental conditions, and processing methods. Analyzing degradation pathways provides valuable insight into the chemical stability of these nanoformulations. 118
Liposomes and niosomes are vesicular drug delivery systems whose stability is governed by the physical integrity of the vesicle, chemical stability of lipids/excipients/drug, and interactions with the environment and storage conditions. 128 Stability assessment combines forced-degradation studies, real-time stability, and accelerated testing with robust analytical methods to monitor the size distribution, encapsulation efficiency, leakage, lipid oxidation, and biological activity. 129
Controlled and Smart Drug Delivery Systems
The stability assessment of advanced drug delivery platforms, such as controlled and smart/stimuli-responsive systems present unique challenges and requires refined strategies compared to conventional pharmaceuticals.
Stability studies for controlled and smart systems follow global ICH Q1A(R2) stability guidelines, which prescribe: Stress, accelerated, and long-term storage at varying temperature and humidity for real-time stability prediction; validation of stability-indicating analytical methods as outlined in ICH Q2 and Q14; Robust documentation and ongoing monitoring, especially for lifecycle updates and change management (Table 4). Lifecycle protocols may evolve as more knowledge is gained, and post-approval monitoring is essential for products with complex release mechanisms.63,130,131
Stability Assessment of Conventional Versus Controlled/Smart Systems.

Novel delivery systems require orthogonal, sensitive, and specific analytical methods to capture both physical and chemical stability. These include HPLC for quantifying drug content and degradation products; MS for molecular characterization of impurities and degradants.; spectroscopic techniques (e.g., IR, NMR) for structural assessment and detection of excipient or matrix instability; Specialized bioassays or in vitro release assays to monitor device or carrier integrity and performance over time, particularly for stimuli-responsive or nanoparticulate systems. 131
Smart drug delivery systems, including stimuli-responsive nanoparticles and hydrogels, must demonstrate both material and formulation stability under the intended storage conditions, as well as stability through potential physiological triggers (pH, redox, light and temperature). In vitro and in vivo compatibility and toxicity testing are critical, as degradation of smart materials can produce byproducts or nanoscale fragments, affecting safety and efficacy. The absence of standardization for many smart systems necessitates the use of tailored protocols and the establishment of device-specific CQAs.132,133
Regulatory Perspectives
The existing guidelines are in the form of individual chapters, leading to variations in their interpretation and application. Also, they do not include modern analytical methods and tools. The recent proposed revisions in the ICH Q1 guidelines mainly aim to have a unified guideline, which was earlier fragmented into ICH Q1A(R2)–Q1E and Q5C. The guideline mainly aims to address stability considerations for advanced therapies and newer methods for stability assessment. The concept article proposes the incorporation of scientifically advanced methods like risk-based approach, stability modeling, predictive tools and modern analytical technologies for stability assessment. The guideline structure has been modernized with detailed annexures, allowing targeted guidelines for specific product types. Annexure 3 is dedicated to ATMP’s, including gene therapies, cell therapies and genome editing tools. These products require specialized stability considerations such as cryopreservation, freeze-thaw cycles and maintenance of viability. This guidance sets them apart from conventional biologic-focused protocols. Annexure 2 introduces scientifically justified stability models and covers linear regression for shelf-life prediction, batch combining methods and scale transformations.
Major organizations such as the International Organization for Standardization (ISO), European Committee for Standardization (CEN), International Electrotechnical Commission (IEC), and ASTM International play a leading role in developing nanotechnology standards. These bodies have formed specialized committees focused on nanotechnology and have collaborated with around 40 countries and research institutions to establish standardized methods for evaluating nanoparticle size and surface modification. The ISO technical report (ISO/TR 18196:2016) consolidates existing techniques for measuring nanomaterial characteristics such as size, morphology, and surface area. Both ISO and ASTM International have published standardization documents covering nearly all analytical methods, except for TERS and AFM-IR, which were later introduced in ISO 18115-2:2021.134,135
Stability studies are typically categorized into three types based on the conditions applied: long-term, intermediate, and accelerated. The stability-evaluation process is often divided into three phases based on the product life cycle. Phase 1 studies are carried out on small-scale laboratory batches and focus on the identification of degradation pathways by simulating stress conditions. Phase 2 involves a comprehensive stability assessment under intermediate and long-term storage conditions. It is carried out on larger-scale technology transfer batches and supports broader clinical studies. Phase 3 studies are aimed at demonstrating the product’s readiness for commercialization. These are full-scale, long-term stability studies that establish shelf life and support regulatory submissions. Multiple production batches are evaluated under operational conditions. 136
Stability studies should involve at least three representative batches, with a minimum of 6 months of data for bulk materials that are stored long-term. Pilot-scale batches may be used initially; however, full-scale batches must be tested post-approval. Batches should reflect the quality of materials used in preclinical and clinical studies, and final-container testing should include different bulk batches, when possible, for comprehensive stability assessment. 21
Challenges and Future Perspectives
During Chemistry, Manufacturing, and Controls (CMC) drug development, creating effective stability programs is often hindered by the limited availability of reliable stability-indicating methods that can differentiate the active drug from its degradation products. Many drugs have intricate structures with multiple functional groups and stereochemical forms, making it difficult to detect and quantify all degradation products. Developing suitable methods requires a detailed understanding of the chemistry and degradation behavior of the molecule. Drugs may degrade through several mechanisms, producing intermediates with unique chemical properties. Identifying and analyzing these intermediates is essential to fully understand the stability characteristics of the drug. Analytical procedures must be highly sensitive and specific to detect even trace levels of degradation products without interference, ensuring the product’s safety, potency, and stability. Stability-indicating methods must be validated for parameters such as accuracy, precision, specificity, and robustness in accordance with regulatory guidelines (e.g., FDA, EMA), which adds complexity and time to the process. Accelerated studies are used to estimate shelf life under stressed conditions; however, identifying appropriate conditions that accurately predict long-term stability remains difficult.
Future perspectives in the stability assessment of NDDS are focused on overcoming the limitations of traditional testing methods using advanced analytical techniques, computational modeling, and artificial intelligence. As NDDS like nanoparticles, liposomes, and micelles grow more complex, new strategies are needed to ensure their quality, safety, and efficacy throughout their lifecycle. 131 Future stability assessment will move toward more sophisticated, high-resolution techniques that can provide real-time, in-depth data on degradation pathways, even at very low concentrations. Techniques such as LC–MS/MS and HRMS enhance sensitivity and resolution, allowing precise detection and characterization of degradation products even at low concentrations. Continuous monitoring, validation, and refinement of analytical methods based on real-time stability data are vital for maintaining their reliability and accuracy.
Conclusion
Stability testing is a crucial aspect of pharmaceutical development, regulatory approval, and lifecycle management, ensuring that drug substances and products consistently maintain safety, quality, and therapeutic efficacy. Although traditional ICH-based protocols offer a solid foundation, their extended timelines and limited predictive capabilities pose challenges in expediting product development. New science- and risk-based technologies, such as APS, ASAP, ASM, and AKM, are revolutionizing stability evaluation by making it a quicker, data-driven, and mechanistically informed process. These methods allow for precise simulation of long-term degradation, more efficient packaging choices, enhanced cold-chain management, and earlier decision-making during development. Strengthening regulatory alignment with modern predictive tools and harmonized standards is essential to fully harness the advantages of NDDSs, biologics, and so on. Overall, integrating traditional frameworks with emerging predictive technologies represents a forward-thinking approach to robust stability programs that reduce development timelines, meet global regulatory expectations, and ensure continuous patient access to high-quality medications.
Footnotes
Acknowledgements
The authors would like to thank MS Ramaiah University of Applied Sciences for providing the necessary facilities.
Authors’ Contribution
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be authors as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
Consent to Participate
Not applicable.
Consent for Publication
All authors have given their consent.
Data Availability Statement
All the data is available with the authors and shall be provided upon request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study does not involve experiments on animals or human subjects.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Not applicable.
Use of Artificial Intelligence-assisted Tools:
The authors declare that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.