
Abstract
Background
Sonodynamic therapy (SDT) has emerged as a promising strategy for cancer treatment; however, its therapeutic efficacy is significantly limited by the hypoxic tumor microenvironment, particularly in breast cancer. To address this limitation, we developed a biomimetic nanoplatform capable of generating oxygen within the tumor microenvironment to enhance SDT performance.
Methods
Poly (lactic-co-glycolic acid) (PLGA) nanoparticles were co-loaded with catalase and the sonosensitizer IR780 and subsequently coated with 4T1 cancer cell membranes (CIP@4T1m NPs). A series of in vitro and in vivo experiments were conducted to evaluate tumor-targeting capability, hypoxia alleviation, singlet oxygen (1O2) generation, antitumor efficacy, induction of immunogenic cell death (ICD), and activation of antitumor immune responses.
Results
The resulting CIP@4T1m NPs were successfully fabricated and exhibited preferential accumulation in 4T1 tumor cells and orthotopic 4T1 tumor-bearing mice. Both in vitro and in vivo studies demonstrated that the nanoplatform partially relieved tumor hypoxia and significantly enhanced SDT-mediated 1O2 production and antitumor effects. Moreover, CIP@4T1m NPs combined with ultrasound induced ICD-associated changes, promoted dendritic cell maturation, facilitated the polarization of tumor-associated macrophages from the M2 to M1 phenotype, reduced regulatory T cell populations, and increased intratumoral CD8+T-cell infiltration.
Conclusion
This work describes a biomimetic nanoplatform that integrates homologous targeting with enzymatic oxygen generation to enhance SDT efficacy and promote antitumor immune responses in an orthotopic 4T1 breast cancer model. The proposed strategy offers a potential approach to mitigate hypoxia-associated limitations in cancer therapy.
Introduction
Breast cancer remains one of the most prevalent malignancies worldwide, with a steadily rising incidence and persistently high mortality, thereby imposing a substantial global health burden. 1 Current clinical management strategies primarily rely on surgery, chemotherapy, radiotherapy, endocrine therapy, and targeted therapy. Although these approaches have substantially improved clinical outcomes, their therapeutic benefits are still limited by tumor heterogeneity, incomplete tumor eradication, treatment resistance, and systemic adverse effects. In particular, conventional chemotherapeutic regimens are frequently accompanied by severe acute and long-term toxicities, which significantly compromise treatment tolerance and patient quality of life. 2 Consequently, the development of therapeutic strategies that combine high antitumor efficacy with improved biosafety remains an important challenge in breast cancer treatment.
SDT has recently emerged as a non-invasive therapeutic modality that leverages the favorable tissue penetration depth and biosafety profile of ultrasound, making it particularly attractive for the treatment of deep-seated solid tumors such as breast cancer. 3 Upon low-intensity ultrasound irradiation, sonosensitizers interact with molecular oxygen to generate 1O2, leading to localized tumor cell damage.4,5 In addition to its direct cytotoxic effects, SDT has been shown to induce ICD, a regulated form of cell death characterized by the exposure or release of damage-associated molecular patterns (DAMPs), including calreticulin (CRT), high-mobility group box 1 (HMGB1), and adenosine triphosphate (ATP). These signals promote dendritic cell (DC) maturation and antigen presentation, thereby facilitating T cell priming and contributing to the conversion of immunologically “cold” tumors into more immune-responsive microenvironments.6,7 Thus, SDT has the potential to function not only as a local tumor-ablation strategy but also as an immune-activating therapeutic approach.
Among the various sonosensitizers investigated to date, IR780 has attracted increasing attention owing to its favorable physicochemical properties and efficient 1O2 generation under ultrasound irradiation. 8 Our previous work demonstrated that IR780-based nanoplatforms can achieve enhanced therapeutic efficacy through multifunctional integration. 9 However, the effectiveness of SDT is often compromised by the hypoxic nature of the tumor microenvironment (TME), which arises from excessive oxygen consumption by rapidly proliferating tumor cells and inadequate oxygen supply due to abnormal tumor vasculature. This hypoxic condition substantially limits 1O2 generation and compromises SDT performance. 9
To overcome hypoxia-associated therapeutic resistance induced by rapid tumor progression, multiple strategies aimed at improving tumor oxygenation have been proposed.10,11 Among them, catalytic decomposition of endogenous hydrogen peroxide (H2O2) to generate oxygen represents a particularly appealing approach, given the intrinsically high H2O2 levels in the TME. 12 Catalase (CAT), a highly efficient antioxidant enzyme, can continuously convert H2O2 into oxygen and water, thereby functioning as an in situ oxygen generator to alleviate tumor hypoxia. Importantly, oxygen availability is a critical rate-limiting factor for SDT, as the generation of 1O2 relies on sufficient molecular oxygen. Therefore, CAT-mediated oxygen production is expected to enhance SDT efficacy by providing an adequate substrate for ultrasound-triggered 1O2 generation. Moreover, CAT-mediated redox modulation has been reported to influence the polarization state of tumor-associated macrophages, suggesting its potential to reshape the immunosuppressive TME. 13 Nevertheless, the clinical application of CAT is hindered by its poor stability and limited intracellular delivery efficiency, highlighting the need for effective delivery systems.
Cell membrane–coated nanoparticles have emerged as a promising class of biomimetic nanocarriers capable of inheriting key biological functions from their source cells, including immune evasion and homologous tumor targeting.14,15 In particular, cancer cell membrane coating preserves tumor-associated antigens and adhesion molecules, enabling selective recognition of homologous tumor tissues and improving tumor accumulation. 16 Such biomimetic camouflage is especially suitable for constructing multifunctional nanoplatforms that require efficient tumor delivery of both therapeutic enzymes and sonosensitizers. Building upon these advantages, we previously reported a 4T1 cancer cell membrane–coated nanoplatform that achieved enhanced therapeutic efficacy through synergistic mechanisms. 17 Therefore, integrating CAT-mediated oxygen generation with IR780-mediated SDT in a 4T1 membrane-coated nanocarrier may provide a rational strategy to simultaneously overcome hypoxia-impaired 1O2 production, improve tumor-selective delivery, and activate antitumor immunity.
In this study, we developed a biomimetic nanotherapeutic system consisting of CAT- and IR780-loaded PLGA nanoparticles cloaked with 4T1 cancer cell membranes (4T1m), which is denoted as CIP@4T1m NPs. The rationale of this design is to integrate three functionally complementary modules into a single nanoplatform: in situ oxygen generation, homologous tumor targeting, and SDT-induced immune activation. Specifically, the PLGA core serves as a biocompatible carrier for the co-encapsulation of CAT and IR780, enabling simultaneous delivery of an oxygen-generating enzyme and a sonosensitizer to tumor tissues. CAT catalyzes the decomposition of endogenous H2O2 in the TME to generate oxygen, thereby alleviating tumor hypoxia and providing sufficient molecular oxygen for IR780-mediated 1O2 production under ultrasound irradiation. Meanwhile, the 4T1 cancer cell membrane coating endows the nanoparticles with homologous tumor-targeting capability and partial immune evasion, which are expected to improve tumor accumulation and reduce nonspecific clearance. Beyond direct tumor cell killing, the amplified SDT effect induces ICD responses, thereby promoting DC maturation and antitumor immune activation. This multifunctional nanoplatform offers an effective strategy for enhancing SDT efficacy while simultaneously modulating the tumor immune microenvironment, as illustrated in Figure 1. Scheme illustration of the fabrication of CIP@4T1m NPs and their antitumor mechanisms.CIP@4T1m NPs were prepared through membrane extraction, a double-emulsion method, and subsequent sonication. Benefiting from the homologous targeting capability of the 4T1 cancer cell membrane, CIP@4T1m NPs preferentially accumulate at tumor sites. Catalase (CAT)-mediated decomposition of endogenous hydrogen peroxide within the tumor microenvironment (TME) generates oxygen, alleviating hypoxia and enhancing singlet oxygen (1O2) production by IR780 under ultrasound irradiation, thereby inducing efficient tumor cell killing. In addition, SDT-induced immunogenic cell death (ICD) triggers the release of damage-associated molecular patterns (DAMPs), which promotes dendritic cell (DC) maturation in tumor-draining lymph nodes and facilitates effector T-cell infiltration into tumor tissues. Consequently, CD8+ T cell -mediated cytotoxicity is enhanced, while immunosuppressive regulatory T cells (Tregs) and M2-like macrophages are reduced, leading to immune remodeling-related changes.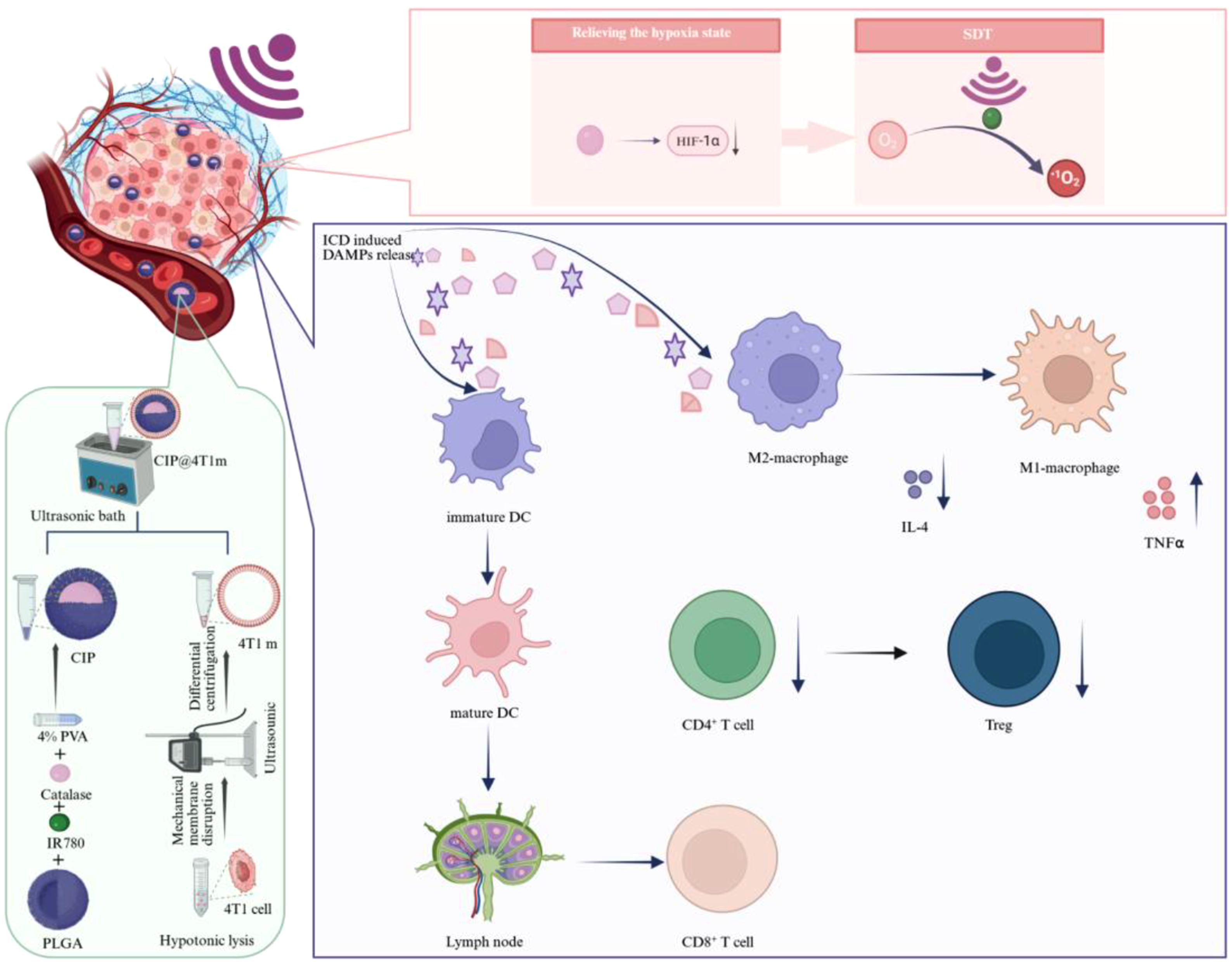
Experimental section
Materials
PLGA, CAT, and poly(vinyl alcohol) (PVA) were obtained from Sigma-Aldrich. [Ru(dpp)3]Cl2 was purchased from Macklin. DAPI, H2DCFDA, and the ATP assay kit were obtained from Beyotime. Fluorescent secondary antibodies were purchased from Proteintech. ELISA kits for TNF-α and IL-4 were obtained from Dakewe. Antibodies for flow cytometry were supplied by BioLegend, while antibodies for CRT, HMGB1, and cytokines were purchased from Thermo Fisher Scientific.
Cell culture
4T1 cells and HUVECs were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. RAW 264.7 cells, bone marrow-derived macrophages (BMDMs), and bone marrow-derived dendritic cells (BMDCs) were maintained in DMEM with the same supplements. All cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Synthesis of CAT/IR780@PLGA nanoparticles (CIP NPs)
CIP NPs were prepared using a water-in-oil-in-water (W/O/W) double-emulsion method with slight modifications from previous reports. 18 Briefly, 50 mg of PLGA was dissolved in 2 mL dichloromethane under magnetic stirring to form a homogeneous organic phase. 4 mg of CAT was dissolved in deionized water (ddH2O). Subsequently, 0.5 mg of IR780 and the catalase solution were added to the PLGA solution under ice-bath conditions, followed by sonication to form the primary emulsion. This emulsion was then poured into 7.5 mL of 4% (w/v) PVA solution and further emulsified using a probe sonicator (40% power, 2 min, 50% duty cycle) under ice-bath and dark conditions. The resulting emulsion was diluted with 40 mL deionized water and stirred for 2–4 h to allow solvent evaporation. Nanoparticles were collected by centrifugation (10,000 rpm, 10 min, 4 °C), washed three times, and stored at 4 °C.
Extraction of 4T1m outer shell
4T1 cell membranes were extracted using a combination of hypotonic lysis, mechanical disruption, and differential centrifugation. Briefly, harvested 4T1 cells were resuspended in ddH2O to induce cell lysis under hypotonic conditions. The resulting cell suspension was then subjected to probe sonication in an ice bath for 4 min to facilitate membrane disruption and release. The lysate was first centrifuged at 5,000 rpm for 5 min to remove nuclei and large cellular debris, followed by centrifugation of the collected supernatant at 15,000 rpm for 30 min to isolate cell membrane fragments. The obtained membrane pellets were collected, and the protein concentration was quantified using a BCA protein assay kit. Purified tumor cell membranes were finally stored at −80 °C for further use.
Preparation of CIP@4T1m NPs
CIP nanoparticles were mixed with 4T1 cell membranes at a mass ratio of 20:9 in deionized water and sonicated to facilitate membrane coating. The resulting biomimetic nanoparticles (CIP@4T1m) were collected by centrifugation and stored at 4 °C in the dark.
Characterization of CIP@4T1m NPs in aqueous solution
The morphology of nanoparticles was examined by TEM. Hydrodynamic size and zeta potential were measured by dynamic light scattering (DLS). SDS-PAGE was performed to compare membrane protein profiles before and after coating. The loading efficiency (LE) and encapsulation efficiency (EE) of IR780 and CAT were calculated separately. For IR780, the amount of unencapsulated IR780 in the supernatant was quantified using UV–vis absorbance according to a standard calibration curve. For CAT, the amount of unencapsulated protein in the supernatant was quantified using the BCA protein assay. The loaded amount was calculated by subtracting the unencapsulated amount from the initially added amount. LE and EE were calculated using the following equations:
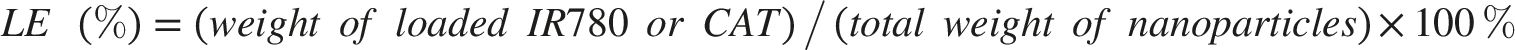
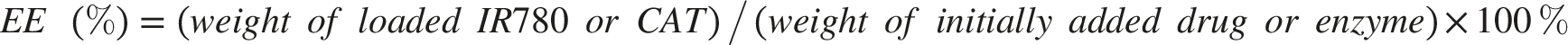
Oxygen generation assay in aqueous solution
IP@4T1m or CIP@4T1m NPs (1 mg/mL) were dispersed in TME-mimicking aqueous solutions (pH 5.5, 100 μM H2O2, hypoxic conditions). The oxygen-generation capability was evaluated by monitoring the dissolved oxygen concentration in each solution at different time points using a portable dissolved oxygen meter, with ddH2O serving as the control.
1O2 generation assay in aqueous solution
IP@4T1m or CIP@4T1m NPs (1 mg/mL) were incubated in TME-mimicking solutions and exposed to ultrasound irradiation (2.0 MHz, 2.0 W/cm2, 20% duty cycle, pulsed mode with 30 s on/off intervals for 2 min). 1O2 generation was subsequently analyzed by electron spin resonance (ESR) spectroscopy using 2,2,6,6-tetramethylpiperidine (TEMP) as the spin-trapping agent.
Cellular uptake
To investigate the cellular uptake of different nanoparticles in tumor cells and macrophages, confocal laser scanning microscopy (CLSM) was used for qualitative analysis. 4T1 cells and RAW 264.7 cells were seeded in glass-bottom confocal dishes at an appropriate density and incubated for 24 h at 37 °C under 5% CO2 to allow cell attachment. The culture medium was then replaced with serum-free medium containing DiO-labeled PLGA, or PLGA@4T1m NPs, followed by incubation for 4 h. After treatment, the cells were washed three times with PBS, fixed with 4% paraformaldehyde for 15 min, and subsequently stained with DAPI to label the nuclei. Fluorescence images were acquired using a CLSM (Leica TCS-SP8, or equivalent) to evaluate the intracellular distribution of nanoparticles. For quantitative analysis, the mean fluorescence intensity of DiO was measured using ImageJ software from at least three randomly selected fields for each group under identical imaging settings.
Cell viability assay
The cytotoxicity of CIP@4T1m NPs was evaluated using a Cell Counting Kit-8 (CCK-8) assay. Briefly, 4T1 cells and HUVECs in the logarithmic growth phase were seeded into 96-well plates at a density of 5 × 103 cells per well and incubated overnight at 37 °C under 5% CO2 in a humidified atmosphere to allow cell attachment. The culture medium was then replaced with serum-free basal medium containing CIP@4T1m NPs at different concentrations (0, 31.25, 62.5, 125, 250, 500, 1000 and 2000 μg/mL), followed by incubation for 24 h. After treatment, 10 μL of CCK-8 reagent was added to each well, and the cells were further incubated for 1–2 h at 37 °C. The absorbance at 450 nm was measured using a microplate reader. Blank wells containing medium and CCK-8 reagent without cells were included for background correction.
In vitro antitumor performance
4T1 cells were seeded in 48-well plates and cultured overnight at 37°C with 5% CO2 until adherence. After removing the medium, each group was treated with PBS, IP@4T1m, CIP or CIP@4T1m for 4 hours. For groups requiring combined ultrasound irradiation, sonication (2.0 MHz, 2.0 W/cm2, 20% duty cycle, 2 min total exposure with 30 s on/off intervals) was applied post-drug incubation. Cells were then stained with Calcein-AM (viable cells) and propidium iodide (PI, dead cells) for 10 min in the dark, followed by gentle PBS washing. Fluorescence images were captured using an inverted microscope, and quantitative analysis of green/red fluorescence signals was performed via ImageJ to determine cell viability and mortality rates for evaluating therapeutic efficacy.
ROS and O2 detection
4T1 cells were seeded in 48-well plates at a density of 2 × 104 cells per well and treated as indicated. Following adherence, cells were subjected to the designated experimental interventions according to the predefined grouping scheme. After 4 hours of treatment, cells were gently washed three times with ice-cold PBS. Subsequently, cells were stained with the oxygen-sensitive probe [Ru(dpp)3]Cl2 (10 μM) or the ROS-sensitive probe DCFH-DA (10 μM) and incubated for 30 min in the dark. Following staining, cells were washed three times with PBS to remove unbound probes. Fluorescence images were captured using an inverted microscope. Quantitative analysis of fluorescence intensity was performed using ImageJ software to evaluate alterations in intracellular oxygen and ROS levels. Reduced [Ru(dpp)3]Cl2 fluorescence was interpreted as increased intracellular oxygen levels, whereas increased DCF fluorescence indicated enhanced intracellular ROS generation.
In vitro ICD induction
Immunofluorescence staining of CRT and HMGB1
4T1 cells were seeded in CLSM-specific dishes and treated according to experimental groups post-adherence. After 4 h of treatment, cells were washed thrice with PBS and fixed with 4% paraformaldehyde at RT for 15 min. For HMGB1 staining, fixed cells were permeabilized with 0.1% Triton X-100 for 10 min to disrupt membranes, followed by blocking with 5% BSA for 30 min. Primary antibodies (anti-CRT or anti-HMGB1) were incubated at RT for 2 h, and corresponding fluorescent secondary antibodies were applied. Fluorescence images were acquired by CLSM. CRT exposure was evaluated by quantifying membrane-associated CRT fluorescence, whereas HMGB1 release was assessed by measuring the reduction of nuclear HMGB1 fluorescence. Quantitative analysis was performed using ImageJ software from at least three randomly selected fields per group under identical imaging settings.
Extracellular ATP assay
4T1 cells were seeded in 6-well plates and treated for 4 h. Culture supernatants were collected and analyzed with an enhanced ATP assay kit according to the manufacturer’s protocol. Luminescence intensity was measured by a microplate reader to quantify ATP release as an indicator of ICD. ATP levels were normalized according to the same sample-processing procedure across all groups.
Isolation and induction of bone marrow-derived immune cells
Bone marrow cells were isolated from femurs and tibias of 6–8-week-old female BALB/c mice by PBS flushing. The collected cells were filtered through a 70-μm cell strainer, subjected to erythrocyte lysis, washed, and resuspended in complete culture medium.
DC maturation assay
To evaluate DC maturation induced by treatment-generated tumor cell signals, immature BMDCs were incubated with supernatants collected from differently treated 4T1 cells for 8 h. After incubation, cells were collected, washed with PBS, and stained with PE-Dazzle594-anti-F4/80, APC-anti-CD80, and PE-Cy7-anti-CD86 antibodies according to the manufacturer’s instructions. Samples were analyzed by flow cytometry (FCM). BMDC maturation was evaluated based on the proportion of CD80+CD86+ cells. FCM data were analyzed using FlowJo software.
Macrophage polarization phenotyping
M2-polarized BMDMs were treated with the indicated formulations or conditioned media for 8 h. After treatment, cells were collected, washed with PBS, and stained with PE-Dazzle594-anti-F4/80, BV711-anti-CD206, and PE-Cy7-anti-CD86 antibodies. Samples were analyzed by FCM. F4/80+ macrophages were gated for phenotypic analysis, and CD86 and CD206 were used as representative markers for M1-like and M2-like macrophages, respectively. The M1/M2 ratio was calculated based on the relative proportions of CD86+ and CD206+ macrophage populations. Data were processed using FlowJo software.
Establishment of In vivo tumor model
Female BALB/c mice (6-week-old, SPF grade) were used for all animal experiments, which were approved by the institutional ethics committee of The Second Xiangya Hospital, Central South University. An orthotopic 4T1 breast tumor model was established by injecting 4T1 cells into the mammary fat pad. Tumor growth was monitored, and treatments were initiated when tumors reached approximately 60 mm3. Tumor volume was calculated as:
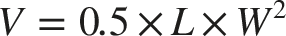
Animals were randomly assigned to different treatment groups. Tumor volume measurements were performed in a blinded manner to minimize potential bias.
In vivo tumor targeting
To evaluate the tumor-targeting distribution of nanoparticles in vivo, near-infrared fluorescent probe IR780 was employed for labeling. Orthotopic 4T1 breast tumor-bearing BALB/c mice were randomly divided into two groups receiving intravenous injections of different nanoparticle suspensions: IR780@PLGA (IP NPs) and IR780@PLGA@4T1m (IP@4T1m NPs). In vivo fluorescence imaging was performed at predetermined time points (4, 8, 12, 24, and 48 h post-injection) using an IVIS Lumina imaging system to monitor whole-body and tumor-specific fluorescence signals. Quantitative analysis of the imaging data was conducted with ImageJ software to assess the accumulation kinetics of nanoparticles in tumor sites. At 48 h post-injection, mice were sacrificed, and major organs (heart, liver, spleen, lungs, and kidneys), tumor tissues, and blood samples were collected for ex vivo fluorescence imaging. The fluorescence intensities of various tissues were quantitatively analyzed using ImageJ software to systematically evaluate the accumulation kinetics of nanoparticles in tumor sites, the biodistribution profile and tumor-targeting capability of the nanoparticles.
In vivo biosafety evaluation
Healthy female BALB/c mice (6-week-old, SPF grade) were randomly assigned to two groups (n=3): saline control group and CIP@4T1m NPs-treated group. The administered dose of CIP@4T1m NPs was normalized to an IR780-equivalent dose of 6.28 mg/kg. This dose was selected based on the measured IR780 loading efficiency of CIP@4T1m NPs, the favorable in vitro cytocompatibility observed at the working concentration, and preliminary tolerability considerations for repeated intravenous administration. The use of an IR780-equivalent dose allowed the amount of sonosensitizer delivered in vivo to be accurately defined and kept consistent with the therapeutic study. The animals received intravenous tail vein injections of corresponding treatments on days 1, 3, 5, 7, and 9. At 24 hours after the final administration, all mice were euthanized by CO2 asphyxiation, and cardiac puncture was performed to collect whole blood samples. Serum was subsequently isolated for comprehensive biochemical analysis, including liver function markers (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) and renal function parameters (blood urea nitrogen [BUN], creatinine [CREA], and uric acid [UA]), to systematically evaluate the in vivo biosafety profile of the nanoparticles.
In vivo anticancer performance
Therapeutic interventions were initiated on day 7 post-tumor inoculation (day 0). Orthotopic 4T1 tumor-bearing mice were randomly allocated into five groups (n=5): saline control, IP@4T1m + US, CIP + US, CIP@4T1m, and CIP@4T1m + US. For all IR780-containing treatment groups, the nanoparticle dose was normalized to an IR780-equivalent dose of 6.28 mg/kg. This dosage was determined according to the experimentally measured IR780 loading efficiency and was chosen to ensure comparable sonosensitizer exposure among different nanoparticle formulations. The selected dose was also supported by the in vitro cytocompatibility results and was further evaluated in the repeated-dose short-term biosafety experiment. Intravenous tail vein injections were administered on days 1, 3, 5, 7, and 9, while ultrasound irradiation (2.0 MHz, 2.0 W/cm2, 20% duty cycle, pulsed mode with 30 s on/off for 2 min) was performed on alternate days (days 2, 4, 6, 8, and 10) for designated groups. Body weight and tumor volume were monitored every 48 hours throughout the treatment period. On day 10, mice were euthanized for collection of major organs (heart, liver, spleen, lungs, and kidneys), whole blood, and tumor tissues. Selected specimens were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned for histological and molecular analyses, including: H&E staining, TUNEL assay, Ki-67 immunohistochemistry, and HIF-1α immunofluorescence. Fresh tumor samples were cryosectioned and subjected to dihydroethidium (DHE) staining for quantitative evaluation of ROS generation.
In vivo immune cell analysis
To evaluate immune cell infiltration within the TME, single-cell suspensions were prepared from murine tumor tissues and tumor-draining lymph nodes (TDLNs). Tissues were mechanically minced, enzymatically digested, and filtered through a cell strainer to obtain single-cell suspensions. After washing with PBS, red blood cells were lysed, and the remaining cells were resuspended for subsequent analysis. Multicolor flow cytometry was employed to characterize different immune cell subsets. Dead cells were excluded using Zombie Aqua™ Fixable Viability Kit where indicated, and immune cell subsets were analyzed within the viable CD45+ cell population.
After staining, all samples were washed with PBS and analyzed by FCM. All FCM data were processed using FlowJo software, and identical gating criteria were applied across different treatment groups to ensure consistency.
Study of immune-related cytokines In vivo
To further evaluate treatment-associated immune-related cytokine changes, key cytokines in serum samples were quantitatively measured using enzyme-linked immunosorbent assay (ELISA). Specifically, the levels of interleukin-4 (IL-4) and tumor necrosis factor-alpha (TNF-α) were determined to assess changes in immunosuppressive and pro-inflammatory cytokine profiles within the experimental observation window. Serum samples were collected from each treatment group, and all ELISA procedures were performed according to the manufacturer’s protocols. Absorbance values were measured using a microplate reader, and cytokine concentrations were calculated based on standard curves generated for each assay.
Statistical analysis
All experiments were performed with at least three independent replicates unless otherwise specified. Quantitative data are presented as mean ± standard deviation. Statistical analyses were conducted using GraphPad Prism. For comparisons among multiple independent groups at a single time point, one-way ANOVA followed by Tukey’s multiple-comparison test was used. For longitudinal tumor volume and body weight data measured repeatedly over time, repeated-measures two-way ANOVA was performed, with treatment and time as the two factors, followed by appropriate multiple-comparison tests. If missing values were present in longitudinal datasets, a mixed-effects model was used. A value of p < 0.05 was considered statistically significant.
Results and discussion
Characterization of CIP@4T1m NPs
CIP NPs were fabricated via a double-emulsion method, in which CAT was encapsulated within the inner aqueous core while IR780 was distributed in the polymer matrix. Subsequently, 4T1 cancer cell membranes were isolated and coated onto CIP nanoparticles to obtain biomimetic CIP@4T1m NPs (Figure 2(a)). SDS–PAGE analysis demonstrated that the protein profile of CIP@4T1m NPs closely matched that of native 4T1 cell membranes, confirming successful membrane coating and preservation of membrane-associated proteins (Figure 2(b)). TEM images revealed a well-defined core–shell structure, with a dense spherical core surrounded by a thin membrane layer (Figure 2(c)). Preparation and characterization of CIP@4T1m NPs. (a) Schematic illustration of the preparation of CIP@4T1m NPs. (b) SDS–PAGE analysis of membrane protein profiles of marker, 4T1 cell membranes (4T1m), and PLGA@4T1m NPs. (c) TEM images of 4T1m, CIP, and CIP@4T1m NPs. (d, e) Hydrodynamic diameter and zeta potential of CIP and CIP@4T1m NPs determined by dynamic light scattering (DLS). (f) ESR spectra for singlet oxygen detection under different conditions. (g) Time-dependent oxygen generation profiles under different conditions. (h) Dispersion stability of CIP@4T1m NPs under different conditions was monitored for 24 h.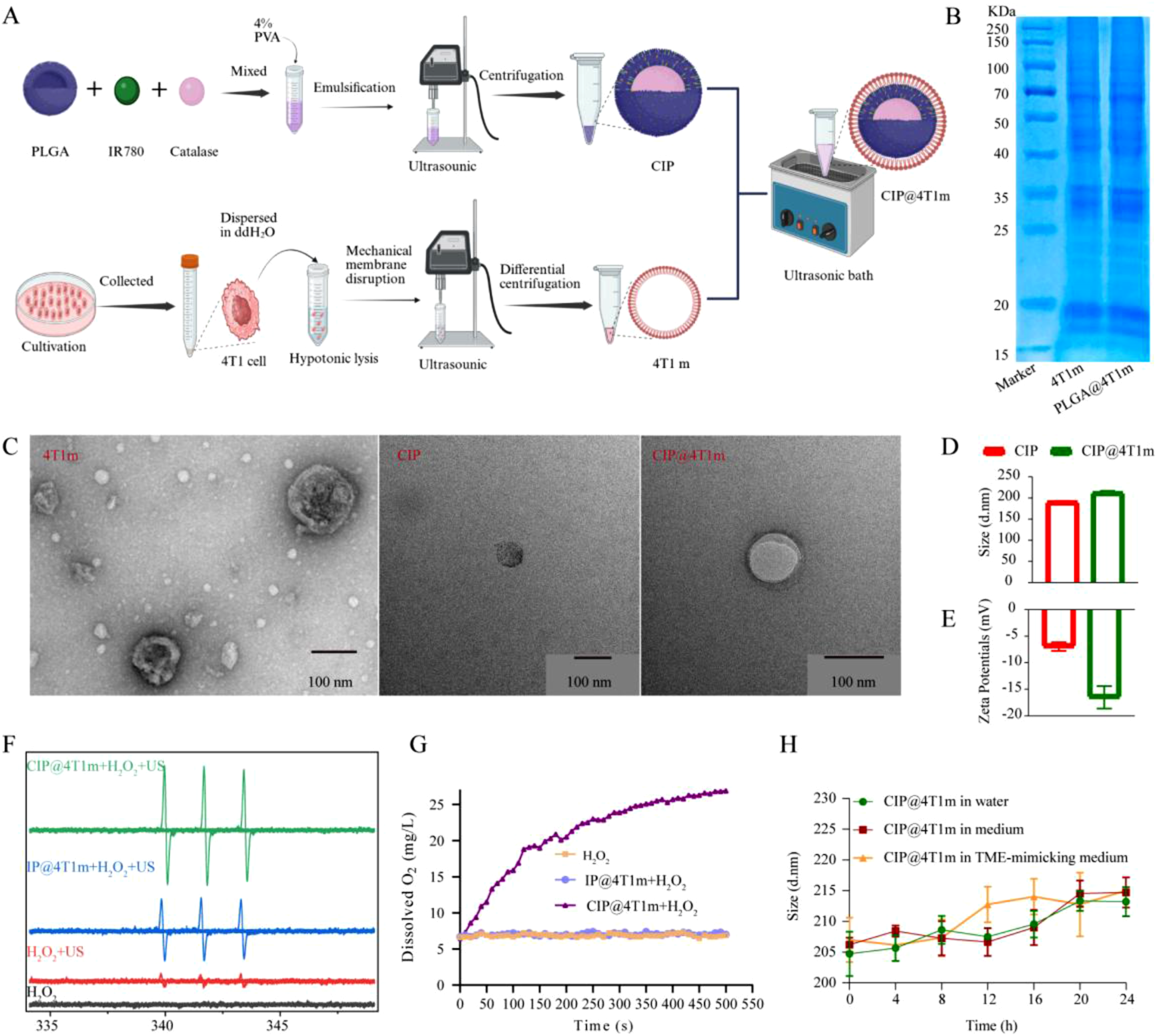
The loading and encapsulation efficiencies of IR780, determined by UV–vis analysis, were 6.28% and 62.8%, respectively. For CAT, the loading and encapsulation efficiencies were 3.25% and 40.62%, respectively, as determined by BCA assay. DLS analysis showed an increase in hydrodynamic diameter from 190.16 ± 2.69 nm to 212.08 ± 4.62 nm after membrane coating, accompanied by a shift in ζ-potential from −6.96 ± 0.81 mV to −16.51 ± 2.12 mV (Figure 2(d) and (e)), suggesting enhanced colloidal stability. Furthermore, negligible changes in particle size were observed over 24 h in deionized water, complete medium, and TME-mimicking solution, demonstrating good stability under physiologically relevant conditions (Figure 2(h)).
The oxygen-generating capability of CIP@4T1m NPs was first evaluated in TME-mimicking conditions. Compared with IP@4T1m, CIP@4T1m exhibited sustained, time-dependent oxygen production, confirming that CAT retained its catalytic activity after encapsulation (Figure 2(g)). Correspondingly, ESR spectroscopy revealed that both IP@4T1m and CIP@4T1m could generate 1O2 under ultrasound irradiation; however, the CIP@4T1m + US group produced markedly stronger signals (Figure 2(f)). This enhancement can be attributed to CAT-mediated decomposition of endogenous H2O2, which continuously supplies oxygen and thereby overcomes hypoxia-imposed limitations on SDT.
Cellular uptake and In vitro biosafety
CLSM demonstrated that 4T1 membrane-coated nanoparticles were preferentially internalized by homologous 4T1 tumor cells, while their uptake by RAW 264.7 macrophages was markedly reduced (Figure 3(a)–(d)). This selective internalization suggests that membrane coating confers both homologous targeting and partial immune evasion, which are advantageous for enhancing tumor accumulation and prolonging systemic circulation in vivo. From a translational perspective, however, certain challenges should be acknowledged. The isolation and preservation of tumor cell membranes require standardized procedures, and issues related to large-scale production and long-term stability remain to be further addressed. In vitro antitumor performance. (a, b) CLSM images of cellular uptake of DiO@PLGA and DiO@PLGA@4T1m NPs in 4T1 cells and RAW264.7 cells. (c, d) Quantification of mean fluorescence intensity (MFI) of DiO (n = 3). Scale bar: 10 μm. (e) Fluorescence images of intracellular oxygen levels using [Ru(dpp)3]Cl2 as a probe after different treatments. (f) Corresponding quantitative analysis of [Ru(dpp)3]Cl2 fluorescence intensity (n = 3). Scale bar: 75 μm. (g) Fluorescence images of intracellular ROS levels using DCFH-DA as a probe after different treatments. (h) Corresponding quantitative analysis of DCF fluorescence intensity (n = 3). (i) Live/dead staining images of 4T1 cells using Calcein-AM (live, green) and PI (dead, red) after different treatments, and (j) their quantitative analysis of cell viability (n = 3). Scale bar: 75 μm. BF: bright field; DiO: green; [Ru(dpp)3]Cl2: red; DAPI: blue; DCF: green. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).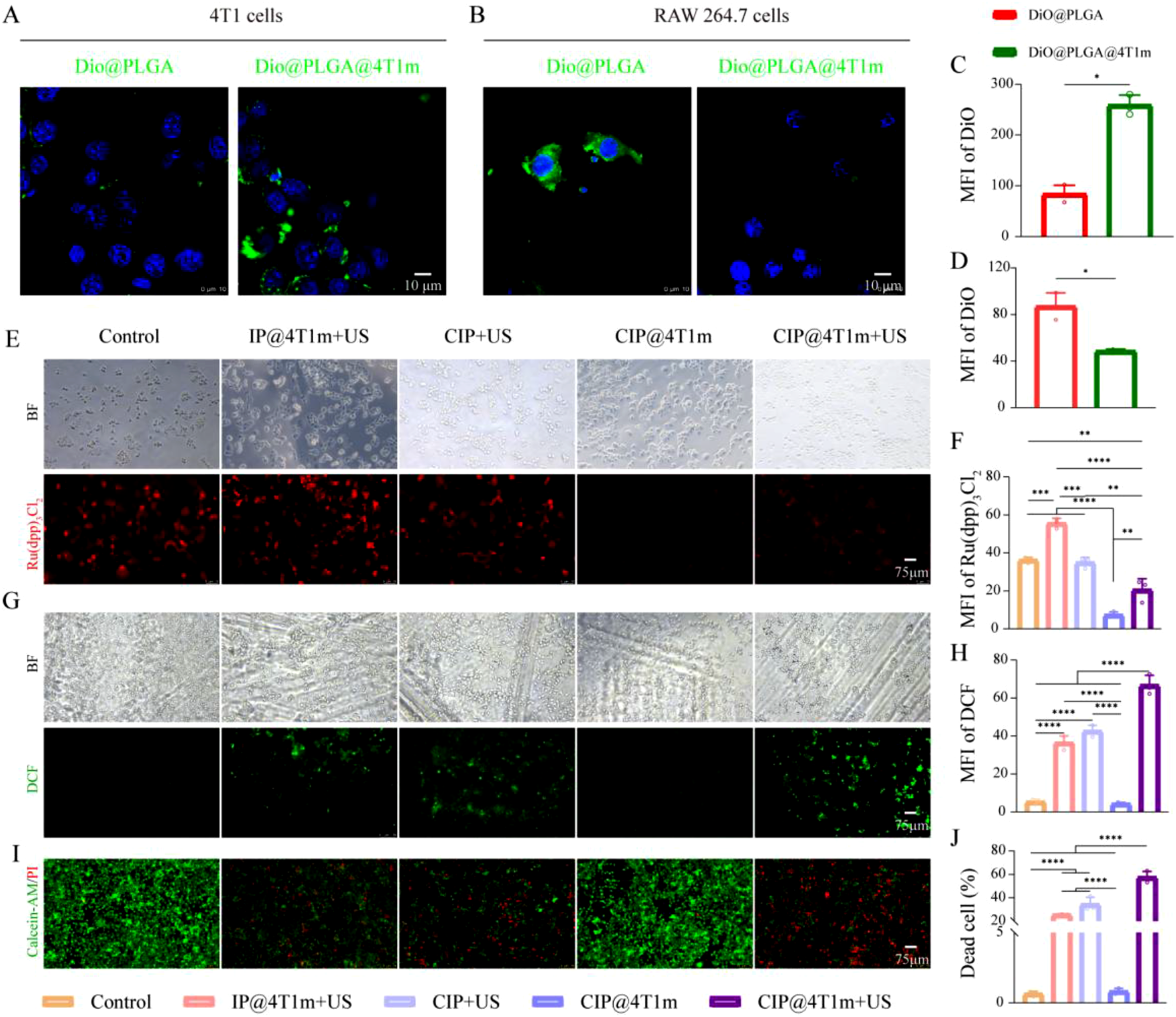
Cytotoxicity evaluation showed that the nanoparticles exhibited minimal toxicity at concentrations up to 1000 μg/mL, whereas moderate cytotoxicity was observed at higher concentrations (Fig. S1A). Based on these results, 1000 μg/mL was selected for subsequent in vitro experiments.
In vitro antitumor activity
Intracellular oxygen levels were evaluated using [Ru(dpp)3]Cl2 as a hypoxia-sensitive probe. The CIP@4T1m group displayed the lowest fluorescence intensity, indicating effective hypoxia alleviation. Upon ultrasound irradiation, fluorescence intensity of [Ru(dpp)3]Cl2 in the CIP@4T1m group increased compared with that of the non-irradiated CIP@4T1m group, although it remained lower than that of the control group. This observation suggests that a portion of the oxygen generated by CAT was consumed during the sonodynamic process for ROS production. (Figure 3(e) and (f)). In contrast, ROS generation was maximal in the CIP@4T1m + US group (Figure 3(g)), which correlated with the strongest cytotoxic effects observed in live/dead staining and CCK-8 assays (Figure 3(i), Fig. S1B). These findings indicate that CAT-mediated oxygen supplementation enhanced ultrasound-triggered ROS generation, thereby improving the in vitro SDT efficacy of CIP@4T1m NPs.
In vitro immunogenic cell death and immune activation
Given the enhanced SDT efficacy of CIP@4T1m + US, we next evaluated whether this treatment could induce ICD. ICD is characterized by the exposure or release of DAMPs, including membrane-translocated CRT, extracellularly released HMGB1, and secreted ATP, which collectively contribute to antigen presentation and antitumor immune activation.
19
Accordingly, CRT exposure, HMGB1 nuclear retention, and extracellular ATP levels were examined after different treatments. The results showed that IP@4T1m + US, CIP + US, and CIP@4T1m + US increased CRT exposure, reduced nuclear HMGB1 retention, and promoted ATP release compared with the control group. Among them, CIP@4T1m + US produced the most pronounced ICD-associated changes, including the highest CRT exposure, the lowest HMGB1 nuclear retention, and the greatest ATP secretion (Figure 4(a)–(e)). These results suggest that oxygen-enhanced SDT promoted ICD induction in 4T1 tumor cells. In vitro antitumor immune activation. (a) CLSM images of CRT exposure on the cell membrane after different treatments. (c) CLSM images showing HMGB1 release from the cell nucleus after different treatments. (b, d) Corresponding quantitative analysis of CRT and HMGB1 fluorescence intensity (n = 3). CRT: red; HMGB1: red; DAPI: blue. (e) Extracellular ATP levels in culture supernatants after different treatments. (f) FCM analysis of CD80+CD86+ mature bone marrow-derived dendritic cells (BMDCs), and (g) their corresponding quantitative analysis (n=3). (h, i) FCM analysis of CD86 and CD206 expression in bone marrow-derived macrophages (BMDMs) after different treatments. (j) Quantitative analysis of the M1/M2 macrophage ratio (n = 3). Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).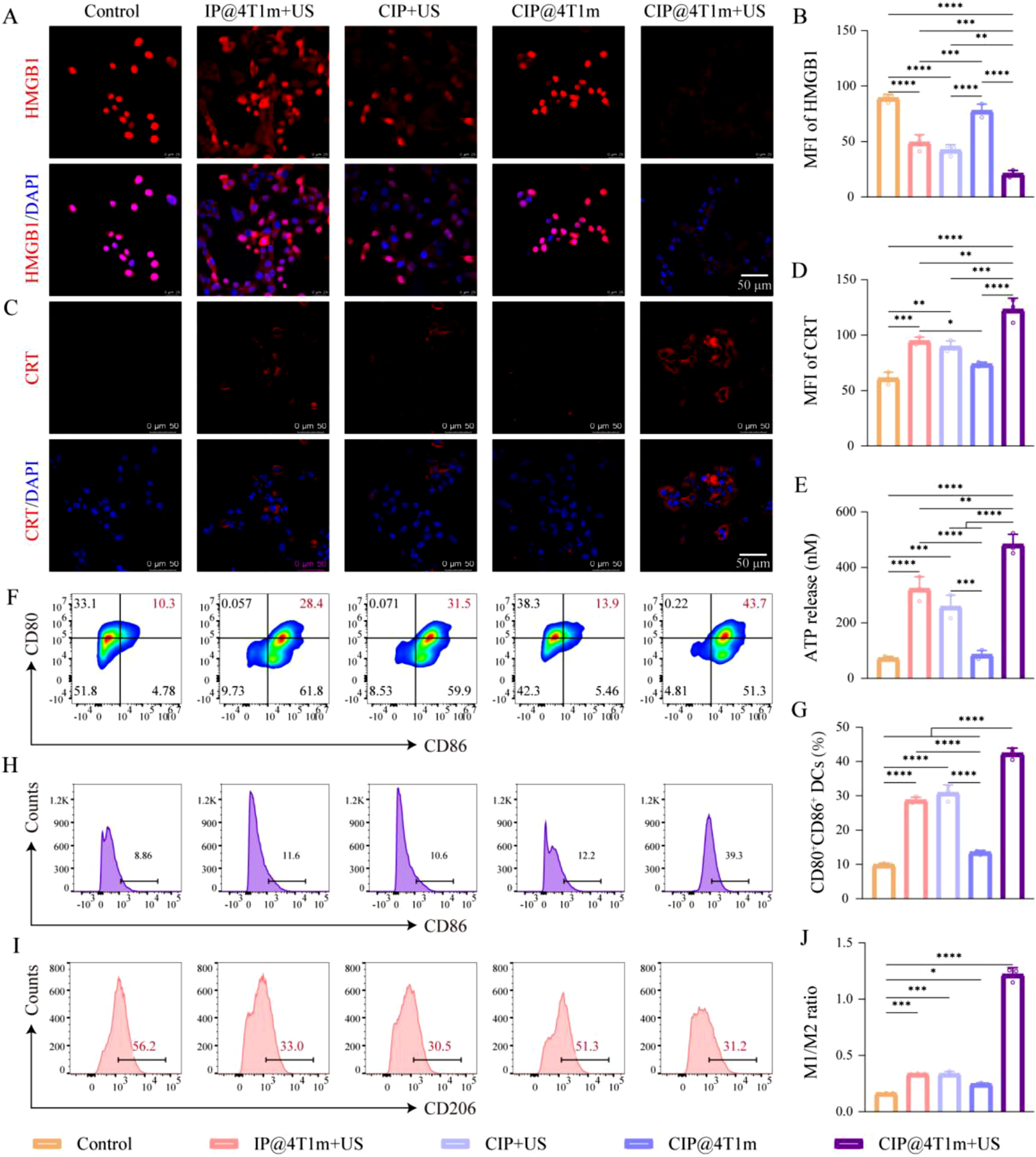
Given the potent ICD-inducing capability of the CIP@4T1m + US strategy, we next investigated its effects on DC maturation and macrophage phenotype repolarization. Mature DCs were identified by the high expression of the costimulatory molecules CD80 and CD86. 20 As shown in Figure 4(f) and (g), BMDCs treated with conditioned medium from the CIP@4T1m + US group exhibited the highest proportion of CD80+CD86+ cells, indicating enhanced DC maturation. Accumulating evidence suggests that ROS can promote the repolarization of pro-tumorigenic M2-like macrophages toward an anti-tumorigenic M1 phenotype within the TME.21–24 High expression of CD86 and CD206 is commonly used to identify M1 and M2 macrophages, respectively. This effect was further examined using BMDMs. CIP@4T1m + US increased CD86 expression and decreased CD206 expression in BMDMs, resulting in the highest M1/M2 ratio among all groups (Figure 4(h)–(j)). Together, these in vitro results indicate that CIP@4T1m + US not only induces ICD but also promotes immune activation and macrophage repolarization.
In vivo targeting and biosafety
To investigate the in vivo tumor-targeting capability of 4T1 membrane-coated nanoparticles, IR780 was employed as a near-infrared fluorescent probe for whole-body imaging. The biodistribution and tumor accumulation of IR780@PLGA nanoparticles (IP NPs) and IR780@PLGA@4T1m nanoparticles (IP@4T1m NPs) were comparatively evaluated. In vivo imaging revealed a time-dependent increase in fluorescence signals at the tumor site in both groups. However, the tumor-associated fluorescence intensity in mice treated with IP@4T1m NPs was consistently higher than that in the IP group, indicating enhanced tumor-targeting ability conferred by the 4T1 cell membrane coating. Notably, the fluorescence signal in the IP@4T1m group reached a maximum at 24 h post-injection and decreased at 48 h. Ex vivo fluorescence imaging performed at 48 h further revealed that the IP@4T1m group exhibited markedly higher IR780 signals in the blood, heart, and tumor tissues compared with the IP group, with the most pronounced accumulation observed in tumor tissues (Fig. S2). These results suggest that 4T1 membrane coating prolongs blood circulation time and improves tumor accumulation efficiency of the nanoparticles. Based on these findings, 24 h post-injection was selected as the optimal time point for subsequent ultrasound irradiation.
Next, the short-term in vivo biosafety of CIP@4T1m nanoparticles was evaluated in healthy mice. The mice were intravenously injected with CIP@4T1m nanoparticles once daily for five consecutive treatments. After completion of the treatment regimen, blood samples were collected to assess liver and kidney function, including AST, ALT, BUN, CRE, and UA. The results showed that there were no significant differences in these biochemical parameters between the CIP@4T1m-treated group and the control group, indicating that CIP@4T1m nanoparticles did not induce noticeable short-term hepatic or renal dysfunction under the tested experimental conditions. (Fig. S3).
Study of in vivo antitumor performance
Based on the tumor-targeting results and short-term biosafety evaluation of CIP@4T1m NPs, their tumor therapeutic efficacy was further evaluated in the orthotopic 4T1 breast cancer model. The treatment regimen is illustrated in Figure 5(a). As shown in Figure 5(b)–(e), after five treatment cycles, tumor growth was significantly inhibited in the CIP@4T1m + US, CIP + US, and IP@4T1m + US groups, whereas the CIP@4T1m group alone showed no notable difference compared with the control group. Among all groups, CIP@4T1m + US exhibited the most pronounced tumor suppression. In vivo antitumor performance. (a) Schematic illustration of the 4T1 tumor-bearing mouse model and treatment schedule. (b) Representative images of excised tumors after five treatment cycles. (c) Tumor volume growth curves during treatment (n = 5). (d) Body weight curves during treatment (n = 5). (e) Individual tumor growth curves for each group. (f) TUNEL staining of tumor sections. TUNEL: red; DAPI: blue. Scale bar: 20 μm. (g) Immunofluorescence staining of Ki67 in tumor tissues. Ki67: red; DAPI: blue. Scale bar: 20 μm. (h) Immunofluorescence staining of hypoxia-inducible factor-1α (HIF-1α). Scale bar: 50 μm. (i) Dihydroethidium (DHE) staining for ROS generation in tumor tissues. Scale bar: 20 μm. For longitudinal tumor volume and body weight curves, statistical significance was analyzed using repeated-measures two-way ANOVA followed by multiple-comparison tests. For endpoint comparisons, one-way ANOVA followed by Tukey’s multiple-comparison test was used.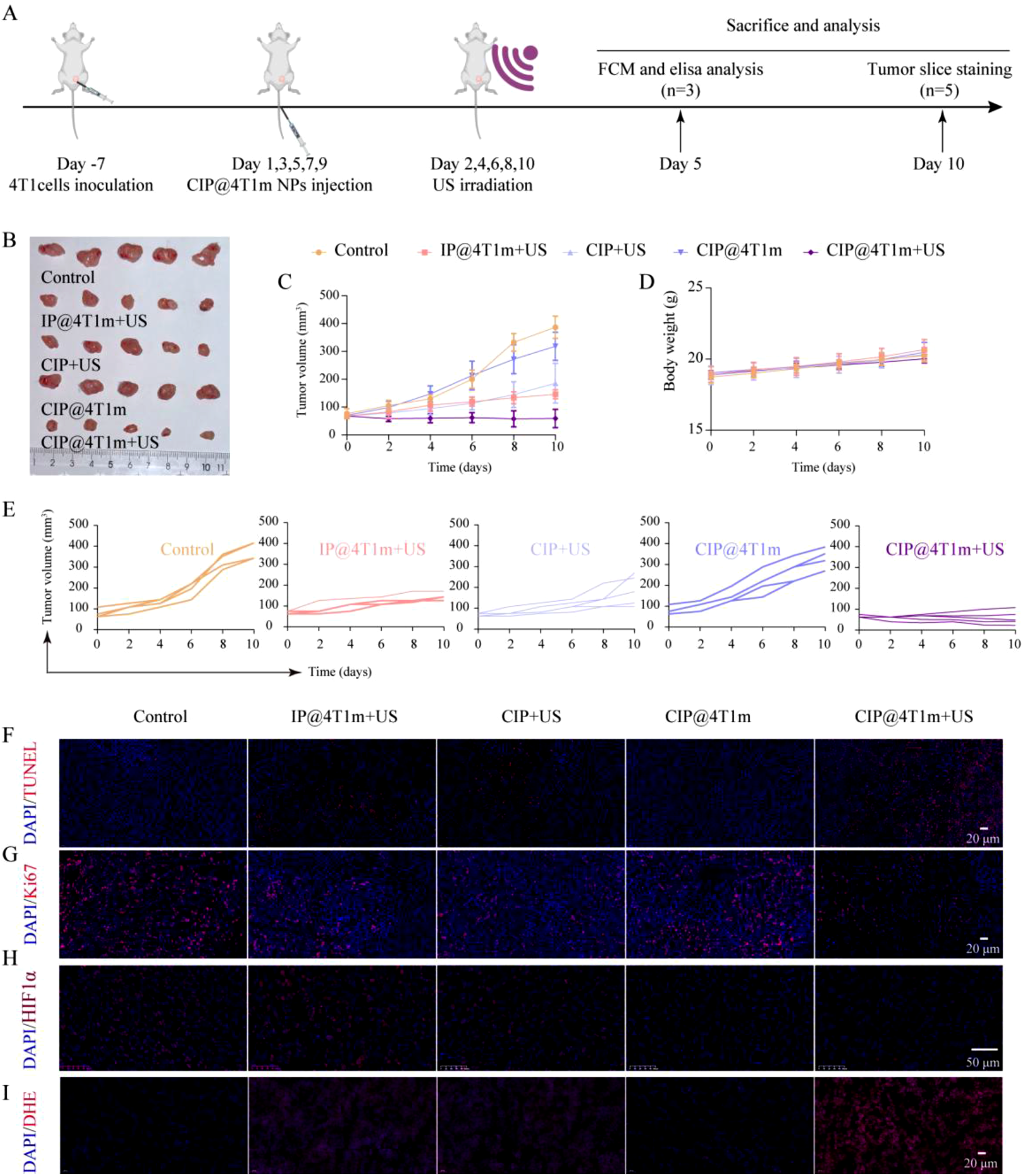
To investigate the underlying antitumor mechanisms, tumor tissues were collected for histological and immunofluorescence analyses, including TUNEL, Ki-67, DHE and HIF-1α staining (Figure 5(f)–(i) and Fig. S4). TUNEL and Ki-67 staining demonstrated that this group exhibited increased tumor cell apoptosis and reduced tumor cell proliferation (Figure 5(f) and (g)).
HIF-1α is commonly upregulated in hypoxic tumor microenvironments. 25 As shown in Figure 5(h), HIF-1α expression was markedly reduced in the CIP@4T1m group, indicating effective hypoxia alleviation by CAT-mediated oxygen generation. In the CIP@4T1m + US group, HIF-1α expression was slightly higher than that in the CIP@4T1m group but remained lower than that in the control group, suggesting that part of the generated oxygen was consumed during the sonodynamic process. Consistently, DHE staining showed the strongest ROS signal in the CIP@4T1m + US group (Figure 5(i)). These results support a coupled mechanism in which CAT-mediated oxygenation improves oxygen availability for ultrasound-triggered ROS production, leading to enhanced tumor cell apoptosis and growth inhibition.
In vivo antitumor immune response
Based on the in vitro immune-activation results and in vivo SDT efficacy of the CIP@4T1m + US strategy, we next evaluated immune-related changes in orthotopic 4T1 tumor-bearing mice. First, TDLNs serve as the primary sites for the activation of antitumor lymphocytes, a process that relies on the efficient delivery and presentation of peripheral antigens.
26
Antigens are unidirectionally drained from the tumor tissue to TDLNs via the lymphatic network, providing a pathway for DCs to actively migrate and carry antigens, thereby initiating immune responses in peripheral tissues.
27
Accordingly, TDLNs were harvested from treated mice, and the maturation of DCs was analyzed by FCM. Mature DCs can present tumor-derived antigens to effector T cells, thereby activating CD8+ T cell-mediated tumor cell killing.
28
Tregs are among the most immunosuppressive populations within the tumor microenvironment and play a central role in tumor progression and immune tolerance.
29
We therefore assessed the infiltration of CD8+ T cells, CD4+ T cells, and Tregs within tumor tissues. Furthermore, we assessed the polarization of tumor-associated macrophages (TAMs) within the tumor tissues. FCM analysis showed that CIP@4T1m + US significantly increased the proportion of CD80+CD86+ mature DCs in TDLNs (Figure 6(a)–(e)). In tumor tissues, this treatment enhanced CD8+T-cell infiltration, reduced Treg populations, and increased the CD8+/CD4+T-cell ratio (Figure 6(b), (c) and (f)–(h)). In addition, CIP@4T1m + US promoted TAM repolarization toward an M1-like phenotype, as evidenced by an increased M1/M2 ratio (Figure 6(d) and (j)–(l)). The gating strategies for FCM analysis are shown in Fig. S5. Serum cytokine analysis further revealed decreased IL-4 and elevated TNF-α levels after CIP@4T1m + US treatment (Figure 6(m) and (n)). These findings indicate that the enhanced local SDT effect was accompanied by enhanced local and peripheral immune responses within the experimental observation window. In vivo antitumor immune response. (a) FCM analysis of CD80+CD86+ mature dendritic cells in tumor-draining lymph nodes (TDLNs). (e) Corresponding quantitative analysis of CD80+CD86+ DCs (n = 3). (b) FCM analysis of CD8+CD3+ and CD4+CD3+ T cells in tumor tissues. (f–h) Corresponding quantitative analysis of CD8+ T cells, CD4+ T cells, and the CD8+/CD4+T-cell ratio (n = 3). (c) FCM analysis of CD4+CD25+FOXP3+ regulatory T cells (Tregs) in tumor tissues. (i) Corresponding quantitative analysis of Tregs (n = 3). (d) FCM analysis of F4/80+CD86+M1-like and F4/80+CD206+M2-like tumor-associated macrophages (TAMs) in tumor tissues. (j–l) Corresponding quantitative analysis of M1-like TAMs, M2-like TAMs, and the M1/M2 ratio (n = 3). (m, n) Serum levels of IL-4 and TNF-α after different treatments (n = 3). Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).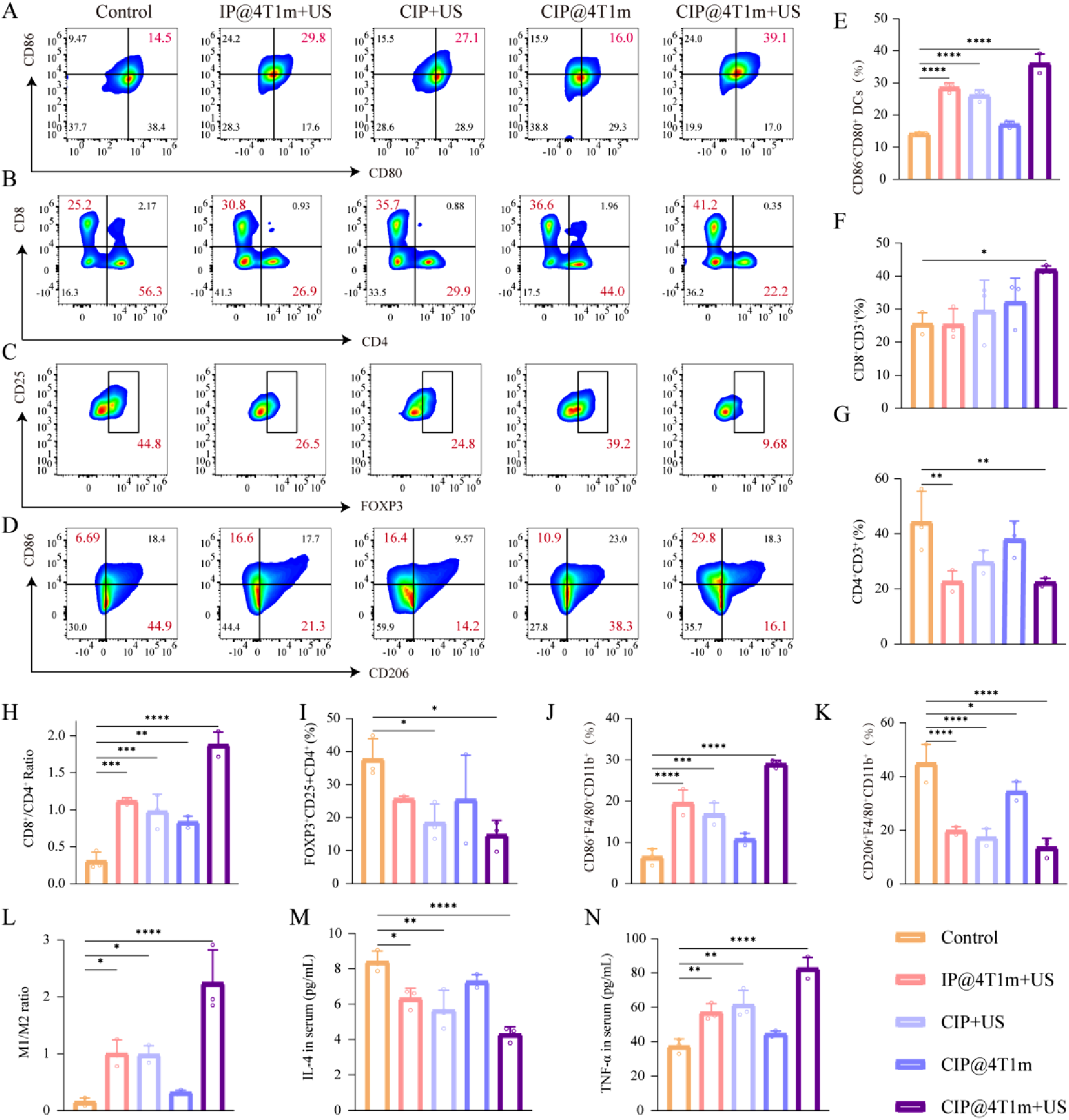
Compared with previously reported oxygen-generating or membrane-coated sonodynamic systems, the present platform combines enzymatic oxygen generation and homologous tumor targeting within one biomimetic nanocarrier. Catalase enables in situ oxygen production from endogenous H2O2, while the 4T1 cell membrane coating improves tumor accumulation and reduces nonspecific uptake. More importantly, the therapeutic benefit observed in this study was not limited to ROS-mediated tumor cell killing, but was further associated with ICD induction and immune microenvironment remodeling. This integrated design provides a practical strategy for enhancing oxygen-dependent SDT under hypoxic tumor conditions.
Conclusion and challenges
In summary, we developed a 4T1 cancer cell membrane-coated PLGA nanoplatform co-loaded with CAT and IR780 for oxygen-enhanced SDT. The resulting CIP@4T1m NPs showed favorable tumor-targeting capability, relieved tumor hypoxia, and improved ultrasound-triggered ROS generation. In the orthotopic 4T1 breast cancer model, CIP@4T1m + US inhibited tumor growth and was associated with ICD induction, DC maturation, TAM repolarization, reduced Treg infiltration, and increased CD8+T-cell infiltration. These findings suggest that oxygen modulation can enhance SDT efficacy and contribute to antitumor immune remodeling in this experimental model.
Despite these encouraging results, several limitations remain. The current biosafety evaluation was limited to short-term biochemical assessment under the tested conditions, and long-term pharmacokinetics, biodegradation, immunogenicity, and chronic toxicity require further investigation.
In addition, this study did not evaluate long-term survival, immune memory, tumor rechallenge, metastatic control, recurrent or postsurgical tumor models, or combination with immune checkpoint blockade. Further validation in additional tumor models is needed to assess the general applicability and translational potential of this approach.
Supplemental material
Supplemental material - A biomimetic oxygen-generating nanoplatform for enhanced sonodynamic immune activation
Supplemental material for A biomimetic oxygen-generating nanoplatform for enhanced sonodynamic immune activation by Haiqin Liao, Shiyu Ding, Ming Zhang, Quanliang Shang, and Zhigang Wang in Journal of Applied Biomaterials & Functional Materials.
Footnotes
Acknowledgments
This project was funded by Joint Project of Pinnacle Disciplinary Group, the Second Affiliated Hospital of Chongqing Medical University (2024201), Hunan Provincial Natural Science Foundation of China (2023JJ30804), and Changsha Natural Science Foundation(kq2208342).
Consent for publication
All authors agree to be published.
Author contributions
Haiqin Liao: Writing – original draft, Writing – review & editing, Methodology, Formal analysis, Data curation. Shiyu Ding: Methodology, Software, and Data curation. Ming Znnhang: Resources. Quanliang Shang: Resources. Zhigang Wang: Guidance, Resources, Supervision, Funding acquisition, Conceptualization. All authors discussed the results and commented on the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Joint Project of Pinnacle Disciplinary Group, the Second Affiliated Hospital of Chongqing Medical University (2024201).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.