
Abstract
The discovery of novel autoantibodies related to idiopathic inflammatory myopathies (collectively referred to as myositis) has not only advanced our understanding of the clinical, serological, and pathological correlation in the disease spectrum but also played a role in guiding management and prognosis. One group of the myositis-specific autoantibodies is anti-aminoacyl-tRNA synthetase (anti-ARS or anti-synthetase) which defines a syndrome with predominant interstitial lung disease, arthritis, and myositis. Autoantibodies to eight aminoacyl-tRNA synthetases have been identified with anti-Jo1 the most common in all of idiopathic inflammatory myopathies. Disease presentation and prognosis vary depending on which anti-aminoacyl-tRNA synthetase antibody is present. In this review, we will discuss the clinical characteristics, overlap features with other autoimmune diseases, prognostic factors, and management of the antisynthetase syndrome.
Keywords
Introduction
Idiopathic inflammatory myopathies (IIM) is a heterogeneous group of systemic autoimmune rheumatic disorders typically characterized by muscular and extra-muscular manifestations particularly skin, joint, and lung of varying severity. The presence of myositis-specific autoantibodies (MSAs) and myositis-associated autoantibodies (MAAs) has become a key feature for classification and diagnosis of IIM and is increasingly used to define clinically distinguishable IIM subsets. 1 Specific MSAs are associated with characteristic clinical phenotypes, which may assist in diagnosis, treatment, and prognostication of IIM and related complications. 2 Among the MSAs, autoantibodies against aminoacyl-tRNA synthetases (ARSs) were detected in 25%–35% of IIM patients. 3 This review will focus on clinical characteristics, overlap features with other autoimmune diseases, prognostic factors, and management of the antisynthetase syndrome (ASSD).
ARS catalyzes the binding of a single amino acid to its specific tRNA during protein synthetase, a process that is ATP-dependent. Autoantibodies to eight ARS have been identified, namely antibodies to Jo-1 (histidyl), 4 PL-7 (threonyl), 5 PL-12 (alanyl), 6 OJ (isoleucyl), 7 EJ (glycyl), 7 KS (asparaginyl), 8 Zo (phenylalanyl), 9 and Ha (tyrosyl). 1 The nomenclature of anti-ARS is based on the name or initials of the index patient. 10 For example, Jo-1 was named after the index patient, John P, a patient with polymyositis (PM) and interstitial lung disease (ILD), in whom anti-Jo-1 was first detected in 1980. 4 Of the eight anti-ARS antibodies, the most common is anti-Jo-1, found in 15%–30 % of patients with IIM11–14 and in 60%–70% of those with ILD. 10 Autoantibodies against the other ARS are less common, each less than 5% prevalence in IIM, however, collectively up to 40% of all ASSD14,15 (Table 1).
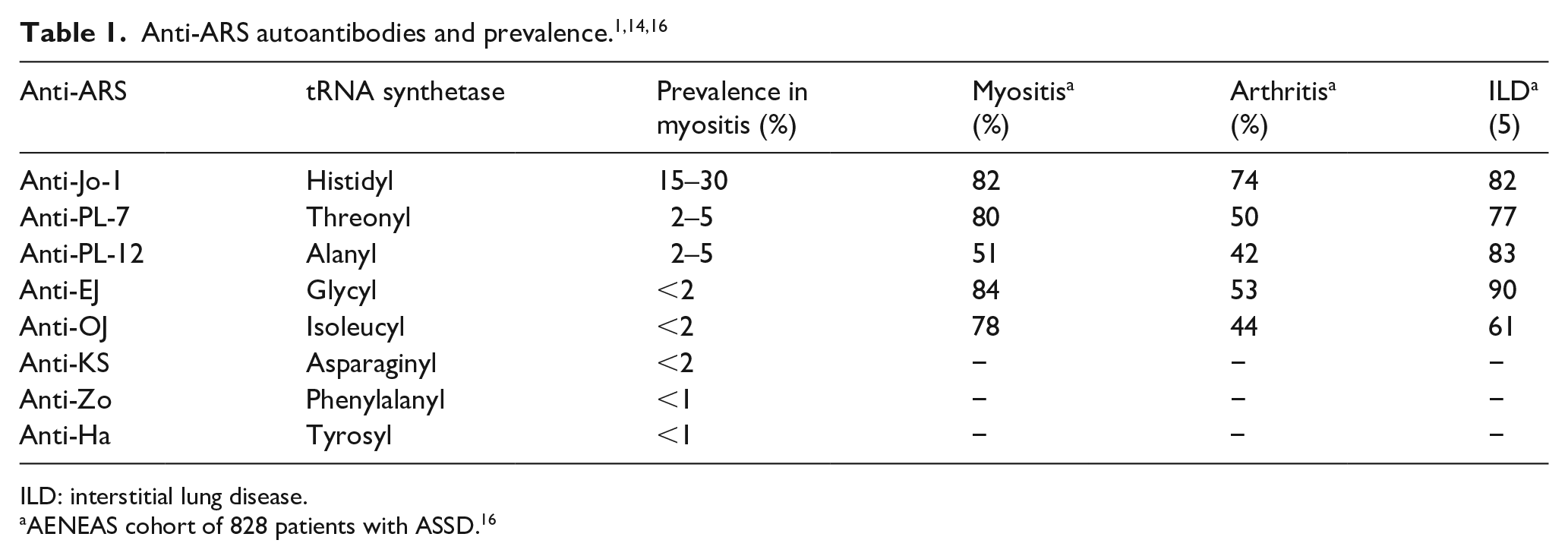
ILD: interstitial lung disease.
AENEAS cohort of 828 patients with ASSD. 16
Although many of the anti-ARS autoantibodies have been shown to inhibit the function of their target autoantigen in vitro, 17 the molecular pathway and the biological significance of the anti-ARS in the pathogenesis of this syndrome remain elusive. Anti-nuclear antibody (ANA) has a poor sensitivity as it is only positive in half of the ASSD patients, and therefore a negative ANA should not be used to exclude this diagnosis. In contrast, ASSD patients frequently (70%–80%) have cytoplasmic staining on indirect immunofluorescence, which may serve as an important screen for ASSD patients in the right clinical setting. 18
Clinical features
The cardinal clinical features of ASSD include myositis, arthritis, ILD, Raynaud’s phenomenon (RP), fever, and mechanic’s hands.15,19–21 Although the typical “triad” of arthritis, myositis, and ILD is observed in up to 90% of cases, 21 clinically it is uncommon to see the concomitant occurrence of all three as initial presentation. Ex novo appearance of clinical features during the follow-up is a typical course of disease evolution in ASSD.21–25 In the American and European NEtwork of Antisynthetase Syndrome (AENEAS) study of 225 patients with anti-Jo1-positive ASSD, only 20% patients had complete triad on presentation. 21 Isolated arthritis, myositis, or ILD occur in up to 50% of cases. Similar findings were reported by a large cohort of Spanish patients with anti-Jo1 ASSD. 26 Sixty percent of the patients with incomplete ASSD developed further manifestations ex novo during the follow-up. 21 At the end of 80-month study period, still 50% of the cohort only had one or two of the triad. 21 Other ASSD typical clinical features, such as fever, RP, and mechanic’s hands, are less frequently observed in comparison with the classic triad findings and have been reported in approximately 40% of cases.15,21,22 Frequency of each manifestations is summarized in Table 2. There is significant heterogeneity in clinical spectrum and time course in ASSD both within the same anti-ARS antibody and among different anti-ARS antibodies. In this section, we will summarize and highlight these unique aspects of ASSD presentation.
Prevalence of anti-Jo-1 ASSD manifestations (%) based on AENEAS cohort data. 21
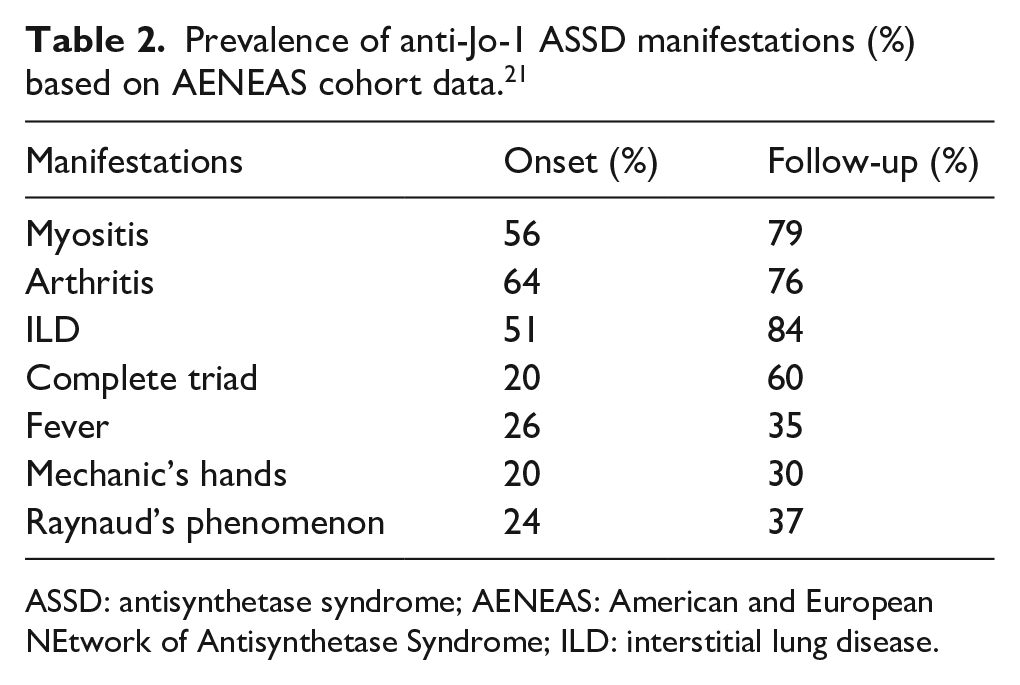
ASSD: antisynthetase syndrome; AENEAS: American and European NEtwork of Antisynthetase Syndrome; ILD: interstitial lung disease.
Arthritis
About 25% of ASSD presents with isolated arthritis alone, and as such, they may be misclassified as having rheumatoid arthritis (RA).22,25,27 In the AENEAS cohort of patients who presented with isolated arthritis, symmetric polyarticular pattern (71%) was the most common, frequently rheumatoid factor (RF)-positive (39%) and/or anti-citrullinated protein antibodies (ACPA)-positive (28%), as well as erosive changes on plain radiograph (35%), 22 making RA a more likely diagnosis in the absence of other ASSD features. Indeed, 70% of these patients actually met the 1987 revised American College of Rheumatology (ACR) classification criteria for RA. 22 Others found that positive ACPA in ASSD was associated with more severe and erosive arthritis, 28 and therefore ACPA positivity in ASSD patients may be considered as a marker of overlap with RA. In these patients, it is important to inquire respiratory and muscle symptoms even during long-term follow-up because up to 90% of patients had ex novo development of either myositis or ILD, or both, in a wide time frame, ranging from a few months to several years. 22
Not all arthritis is the same. Arthritis characteristics are heterogeneous within the syndrome and influenced by its timing of onset. Patients who have arthritis at onset of ASSD commonly have symmetric polyarthritis (70%), whereas those with ASSD who present with ex novo arthritis during follow-up are much more frequently oligoarticular and asymmetrical, more likely to have ASSD accompanying findings, that is, fever, RP, and mechanic’s hands, and less likely to have RA-like clinical, laboratory, and erosive radiographic features. 23
Myositis
Although ASSD is typically described as a subset of inflammatory myositis, muscle involvement is not the most frequent finding at first presentation.21–24 At the onset of ASSD, only 23.5% in the Pittsburgh cohort 14 and 52% in the AENEAS cohort 29 had muscle involvement. At the end of study follow-up, still about 25% in the Pittsburgh cohort 14 and 20% in the AENEAS cohort were amyopathic. 21 In contrast, much higher rate of myositis (90%) was reported in the ASSD subset of the EuroMyositis registry, in which muscle involvement was the common reason for inclusion. 30 The clinical pattern of muscle involvement in ASSD is typical of IIM with subacute to chronic progressive proximal greater than distal symmetrical muscle weakness with elevated muscle enzyme and myopathic electromyography (EMG), therefore often initially diagnosed as PM. However, about 20%–25% ASSD patients have DM rashes with other typical features of myositis and therefore are clinically diagnosed as DM.
There is a considerable clinical heterogeneity among ASSD with different subtypes of anti-ARS antibodies. Jo-1 patients are much more likely to have muscle involvement than non-Jo-1 patients. 14 A retrospective analysis of 166 adult Japanese patients with anti-ARS antibodies showed that myositis was well represented in those with anti-Jo-1, anti-EJ, and anti-PL-7 both at disease onset and end of follow-up period, whereas most of those with anti-PL-12, anti-KS, and anti-OJ are amyopathic throughout the entire clinical course. 31
ILD
ILD is a hallmark feature of ASSD. The overall prevalence of ILD varies from 70% to 95% among ASSD patients depending on the cohorts.21,30,32 For example, in the AENEAS cohort of anti-Jo1 patients, 50% had ILD at disease onset and 84% at the end of the 80-month follow-up with three main patterns, acute/subacute, chronic, and asymptomatic, each about 1/3. 21 The overall prevalence of ILD reached 94.4% in a Chinese ASSD cohort after a follow-up of 22 years. 32 Rate of rapid progressive (RP) ILD was reported as high as 10% in this cohort. 32 Similarly, in a Japanese cohort of 166 patients with ASSD, almost all eventually suffered from ILD with 7.8% RP-ILD at their first visit or during their clinical course. 31 The higher prevalence of RP-ILD in Asian studies compared to Western cohorts suggests perhaps a racial difference in the pulmonary manifestation of this entity. ILD has been shown to confer high mortality and morbidity in the ASSD patients.33,34 In the Pittsburgh ASSD cohort, 49% of mortality was attributed to ILD. 14 Given high frequency and mortality associated with ILD, patients with ASSD should be screened for ILD with CT chest even if asymptomatic clinically.
There is heterogeneity among different anti-ARS antibodies in prevalence and severity of ILD. Patients with anti-PL-7 and anti-PL-12 autoantibodies more frequently have ILD 34 and less likely have myositis compared to anti-Jo-1 patients. 20 RP-ILD occurs more in the non-Jo-1 patients, particularly in those with coexisting anti-PL-7 and anti-Ro-52. 32 Non-Jo-1 patients had a significantly worse unadjusted cumulative and event-free survival from diagnosis than Jo-1 patients. 14 Diagnosis in non-Jo-1 patients is often delayed due to atypical or subtle presentation of connective tissue disease (CTD) features, which negatively affects their survival. 14
High-resolution computed tomography (HRCT) of chest commonly reported bibasilar fibrosis, ground-glass opacities, interlobular reticulation, and traction bronchiectasis.35,36 Although non-specific interstitial pneumonia (NSIP) is the most common radiographic pattern followed by organizing pneumonia (OP) and usual interstitial pneumonia (UIP) in patients with ASSD associated ILD, correlation with histopathology is poor. Histologically, UIP is the most common form of ILD in anti-Jo-1, 37 anti-PL-7, 38 and anti-PL-12 cohorts. 39 No pathognomonic features that would allow for a definitive diagnosis of a particular ARS based on the histopathologic features alone. 39
Most patients with ASSD who had myositis without ILD at the onset of disease will eventually develop ILD later given the high frequency of ILD associated with ASSD. 31 It is also important to recognize that 25% of patients with ASSD had ILD alone at disease onset and may never evolve to have muscle involvement or myositis or other characteristics. 31 In other words, ILD alone could be the only clinical signs of ASSD.31,40 Except for anti-Jo-1, all the other anti-ARS antibodies are not routinely tested in the autoimmune serology panel. As a result, many patients with non-Jo-1 ASSD would otherwise be classified under idiopathic pulmonary fibrosis (IPF) or interstitial pneumonia with autoimmune features (IPAF), thus potentially entering in clinical trials addressed to other conditions or not getting appropriate treatment for ASSD. This point was illustrated by Scire et al., 24 who showed 36% of patients with anti-ARS antibodies who initially met the criteria for IPAF ultimately evolved and reclassified as IIM. Many experts now suggest that patients with ILD, especially with atypical findings on CT for a classic IPF, should be tested for anti-ARS antibodies. 41 Indeed, anti-ARS antibodies were found in 6%–25% of patients with IPF, many of whom had no extra-pulmonary features.42–45 Whether or not skin or muscle involvement is evident, patients with anti-ARS antibodies exhibit similar ILD pattern and response to immunosuppressants. 41 These patients are important to recognize, as a diagnosis of myositis-associated ILD, even for same histological subtype of UIP, is associated with a considerably better prognosis than IPF. 46 These findings further emphasize the importance of establishing clinicoserological classification criteria for ASSD.
Pulmonary artery hypertension
Pulmonary artery hypertension (PAH) was found in 11% of Jo-1 and 21% of non-Jo-1 ASSD patients in the Pittsburgh myositis cohort, 14 either by right heart catheterization or echocardiography. Pulmonary hypertension is the second most common cause of death (11%) in this cohort. 14 A retrospective French study noted pulmonary hypertension in one-quarter of ASSD patients by echocardiography; 8% of the cohort was confirmed severe PAH by right heart catheterization. 47 This was likely an underestimate as only selected few underwent right heart catheterization. Presence of pulmonary hypertension was significantly associated with a lower survival rate, with a 3-year survival rate of 58%. 47 Pulmonary hypertension is under-recognized in patients with ASSD. We recommend doing screening echocardiogram at baseline even for asymptomatic individuals with ASSD and subsequently on follow-up for any evidence of exercise-induced desaturation or dyspnea. 48
Other accompanying features
Mechanic’s hand represents the typical cutaneous manifestation of ASSD. 49 The novel finding of Hiker’s feet is the equivalent of mechanic’s hands, newly recognized in the recent years.50,51 These are characterized by hyperkeratotic scaly fissured plaques on the fingers/palms or soles, generally more evident on the lateral aspect of index fingers.52,53 Mechanic’s hand is suggestive of but not specific to ASSD and can be found in IIM associated with other autoantibodies.31,54 Presence of mechanic’s hand in ASSD is associated with a higher rate of ILD in some studies.53,55,56
RP is another non-specific symptom associated with ASSD although the exact mechanism of vasculopathy in ASSD is unclear. RP is more prevalent in ASSD associated with PL-7 and PL-12 in comparison with Jo-1, 57 and rarely, cases of recurrent digital ulcers have been reported. 58 Nailfold video capillaroscopy (NVC) abnormality can be seen in more than 60% of patients with ASSD. 59 The presence of an systemic sclerosis (SSc)-like pattern is common in ASSD especially with anti-Jo-1 antibody but is independent from the occurrence of RP, 59 suggesting nailfold exam perhaps should be a routine part of ASSD evaluation even without RP symptoms.
Other than the classic features mentioned above, calcinosis, panniculitis, coronary artery dilatation, eosinophilic pleural effusions, and myopericarditis have also been reported in small case series.60–69
Overlap with other CTDs
Overlap between IIM and other CTD is common. The immunological profile is generally characterized by the presence of autoantibodies which are not specific to but frequently associated with myositis. Anti-PM/Scl, anti-Ku as well as anti-U1 RNP and anti-SSA/Ro represent the most frequent MAAs. The analysis of seven large cohorts in the literature showed that 6.5%–36.7% of the patients with IIM presented overlap syndrome,70–75 most commonly SSc, approximately 30%–50%70,76,77 also known as scleromyositis. In SSc cohort, about 6%–8% have SSc-myositis overlap.78,79
However, ASSD overlap with SSc is not commonly reported and has been shown only in a few case reports in PL-7, PL-12, and Jo-1.70,80 In a Dutch SSc cohort, only 2 of the 422 patients with SSc patients had Jo-1 antibody. 78 Although the pathogenesis of SSc and ASSD seems to be quite different, vasculopathy is a typical manifestation shared by both diseases. De novo development of sclerodactyly, RP, and finger-tip ulcerations with positive RNP antibody was reported in a case of established anti-PL12 ASSD. Scleroderma-like renal crisis with biopsy evidence of thrombotic microangiopathy was described in one case of new diagnosis of anti-PL-7 ASSD who had negative usual SSc antibodies (ENA, RNA polymerase III, Scl, and centromere). 81
Histopathological features of the muscle biopsy
Clinico-sero-pathological correlation is increasingly recognized. ASSD is a distinctive pathological subset among IIM. The hallmark of ASSD in muscle pathology is perifascicular necrosis (necrosis and regenerating fibers located at the periphery of the muscle fascicle), 82 although diffusely distributed necrotic and regenerating fibers not restricted to the perifascicular area have also been described in a large cohort of 50 ASSD patients. 83 Very frequently, there is increased expression of major histocompatibility complex (MHC) class I and class II in the cytoplasm and sarcolemma fibers and sarcolemmal membrane attack complex deposition, predominantly in the perifascicular region.83–86 Other findings also include perimysial connective tissue fragmentation and alkaline phosphatase overexpression in perimysium.83,87,88 ASSD muscle biopsy can be sometimes be misclassified as dermatomyositis (DM), which is characterized by the presence of perifascicular inflammation, myofiber atrophy, MHC upregulation,89,90 and immune-mediated necrotizing myopathy (IMNM), which is characterized by diffuse muscle necrosis with little to no endomysial lymphocytic infiltration. 89
Anti-Ro 52 in ASSD
Of the MAAs, anti-Ro 52 antibody draws much attention with regard to its importance in both prognosis and therapeutic implication. The cytoplasmic Ro/SSA antigen has two polypeptide components: Ro 52 and Ro 60, autoantibodies to both of which are commonly seen in CTD. However, Ro 52 is biochemically and immunologically distinct from Ro 60 and is considered more immunogenic. 91 Anti-Ro 52 is present in about 30% of IIM 92 and has a strong association with anti-ARS antibodies in comparison with other MSAs or MSA negative DM.1,93,94 It was found in 40%–72% of patients with anti-Jo-1 ASSD and up to 70% in those with non-Jo-1 ASSD depending on the population studied, and there was no associated concomitant Sjögren syndrome clinically.11,93–97 Some reported that patients with anti-PL7, anti-PL12, and anti-EJ were much more likely to have anti-Ro 52 than those with anti-Jo-1. 32
Presence of anti-Ro 52 in patients with ASSD has been related to more severe ILD, relapses, and refractory disease97–99; however, correlation of this autoantibody with degree of myositis, arthritis, or skin manifestations is inconsistent between studies.95,100 Acute onset respiratory failure and development of lung fibrosis was more frequently observed in ASSD with anti-Ro 52 than those without anti-Ro 52, particularly in anti-PL-7 positive patients, 32 with high anti-Ro 52 concentrations showing the highest risk. 100 Similar effect of anti-Ro 52 on ILD has also been reported in association with other MSAs particularly anti-MDA5. 101
At least in anti-Jo-1 ASSD, presence of high-concentration anti-Ro 52 also confers significance in therapeutic response. Bauhammer et al. showed patients with high anti-Ro 52 titer responded poorly to conventional immunosuppressants including cyclophosphamide (CYC) and cyclosporin but all responded well to rituximab (RTX). In contrast, those with low-titer anti-Ro 52 did well on conventional immunosuppressants alone.95,100
The mechanism as to why concomitant anti-Ro 52 with anti-ARS leads to more severe clinical phenotype remains elusive. Ro 52 is also known as TRIM21 (tripartite motif proteins). It is an interferon (IFN)-inducible E3 ligase that mediates the ubiquitination and proteasomal degradation of various IFN regulatory factor transcription factors, leading to a downregulation of Type-I IFN. 102 It is postulated that antibody to Ro 52 suppresses the degradation and therefore excessive production of proinflammatory cytokines. 102
Malignancy in ASSD
Previously, a negative association between ILD, especially in ASSD patients, and cancer was accepted. In a large retrospective analysis of 233 patients with ASSD, the frequency of cancer was only 1.7% within 3 years from ASSD diagnosis, in which the authors assert was not much different than the general population in France. 20 However, other case studies and review demonstrated a significant increased risk of cancer in these patients, ranging from 4% to 13% of ASSD patients.26,32,103,104 Additional cases and small series have also been published.19,20,105–113 Most frequently identified cancer types are lung and colon, and less frequently breast and ovarian. 105 Only one study reported association of positive anti-Ro 52 with higher prevalence of malignancy. 95 Malignancy is associated with poor prognosis in ASSD. 32 These emerging data support that even though overall cancer risk may be lower than other autoantibody subsets, still a diligent malignancy screen for ASSD patients is warranted.
Outcomes in ASSD
Some negative prognostic factors have been recently identified in ASSD. The presence of anti-Ro 52, RP-ILD, PH, and malignancy have been identified to associate with poor prognosis. 32 Others showed male sex, reduced DLCO (diffusing capacity of lung for carbon monoxide) at presentation, and elevated serum ferritin correlated with reduced survival.32,36,114 Black American ethnicity is also an independent prognostic factor associated with increased lung involvement severity. 115
Reported survival rate of ASSD patients are fairly similar between various studies.14,26,32,36 The Pittsburgh myositis registry of 202 ASSD patients followed over 25 years (1985–2009) showed overall 33% mortality and 6% required lung transplantation. The 5- and 10-year unadjusted cumulative survival rates from diagnosis were 84% and 61%, respectively. 14 Analysis from a large Chinese cohort of 128 ASSD patients showed cumulative 10-year survival rate of 76.8% 32 and similarly, the Spanish cohort of 148 anti-Jo-1 ASSD patients had 25% mortality after a median follow-up of 6 years. 26
Among different anti-ARS antibodies, anti-non-Jo-1 ASSD have worse clinical outcome from anti-Jo-1 patients.14,116,117 The anti-Jo-1 patients had a significantly better 5- and 10-year unadjusted cumulative survival from diagnosis (90% and 70%, respectively) as compared to non-Jo-1 patients (75% and 47%, respectively). 14 Several variables affect the survival difference between anti-Jo-1 and non-Jo-1 patients. A major factor is the delay in diagnosis as anti-non-Jo-1 patients often present atypically with more frequent ILD 34 and non-specific presentation, and less frequently myositis or arthritis. 20 Second, anti-non-Jo-1 antibodies are not routinely tested or available, and when tested, the reliability of commercial testing is highly variable. Finally, anti-non-Jo-1 ASSD, particularly anti-PL7, is associated with more frequent RP-ILD which confers poor prognosis. 32
Treatment in ASSD: focus on ILD
Managing ASSD represents a challenge to the clinicians as patients can have multi-system involvement, ILD being the most life-threatening. Muscle, skin, joint, and lung disease activity in ASSD may have different clinical course and response to therapy and often require multidisciplinary approach from rheumatologists, dermatologists, and pulmonologists. In Table 3, we recommend to thoroughly review patients and arrange investigations by organ systems at first visit and during each follow-up.
Recommended measures and investigations at diagnosis and each follow-up.

TJC: tender joint count; SJC: swollen joint count; PAH: pulmonary artery hypertension; MMT8: manual muscle testing 8 (https://www.niehs.nih.gov/research/resources/assets/docs/mmt8_grading_and_testing_procedures_for_the_abbreviated_8_muscle_groups_508.pdf); MRI: magnetic resonance imaging; EMG: electromyography; CXR: chest X-ray; HRCT: high-resolution computed tomography; PFT: pulmonary function test; 6MWT: 6-minute walk test; DLCO: diffusing capacity of lung for carbon monoxide; ILD: interstitial lung disease; CDASI: cutaneous dermatomyositis disease area and severity index; CBC: complete blood count; CK: creatine kinase; LDH: lactate dehydrogenase; AST: aspartate aminotransferase; ALT: alanine transaminase; ANA: antinuclear antibody; ENA: extractable nuclear antigens; RF: rheumatoid factor; ACPA: anti-citrullinated protein antibody; MSA: myositis-specific antibody; MAA: myositis-associated antibody; MDAAT: Myositis Disease Activity Assessment Tool; MDI: myositis damage index; HAQ: Health Assessment Questionnaire; SF-36: Short-Form 36 Health Survey Questionnaire.
ILD is a major cause of morbidity and mortality in myositis requiring combinations of glucocorticoids, immunosuppressive drugs, and agents that modulate T-cell function and deplete B-cells. Unlike the antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis literature, there is little evidence in choice of drugs or duration of therapy in “induction” and “maintenance.” There are very few large treatment trial results available to guide myositis treatment and none for myositis-associated ILD. We will focus the discussion on myositis-associated ILD treatment with emphasis on ASSD in this section. A proposed treatment algorithm is illustrated in Figure 1.
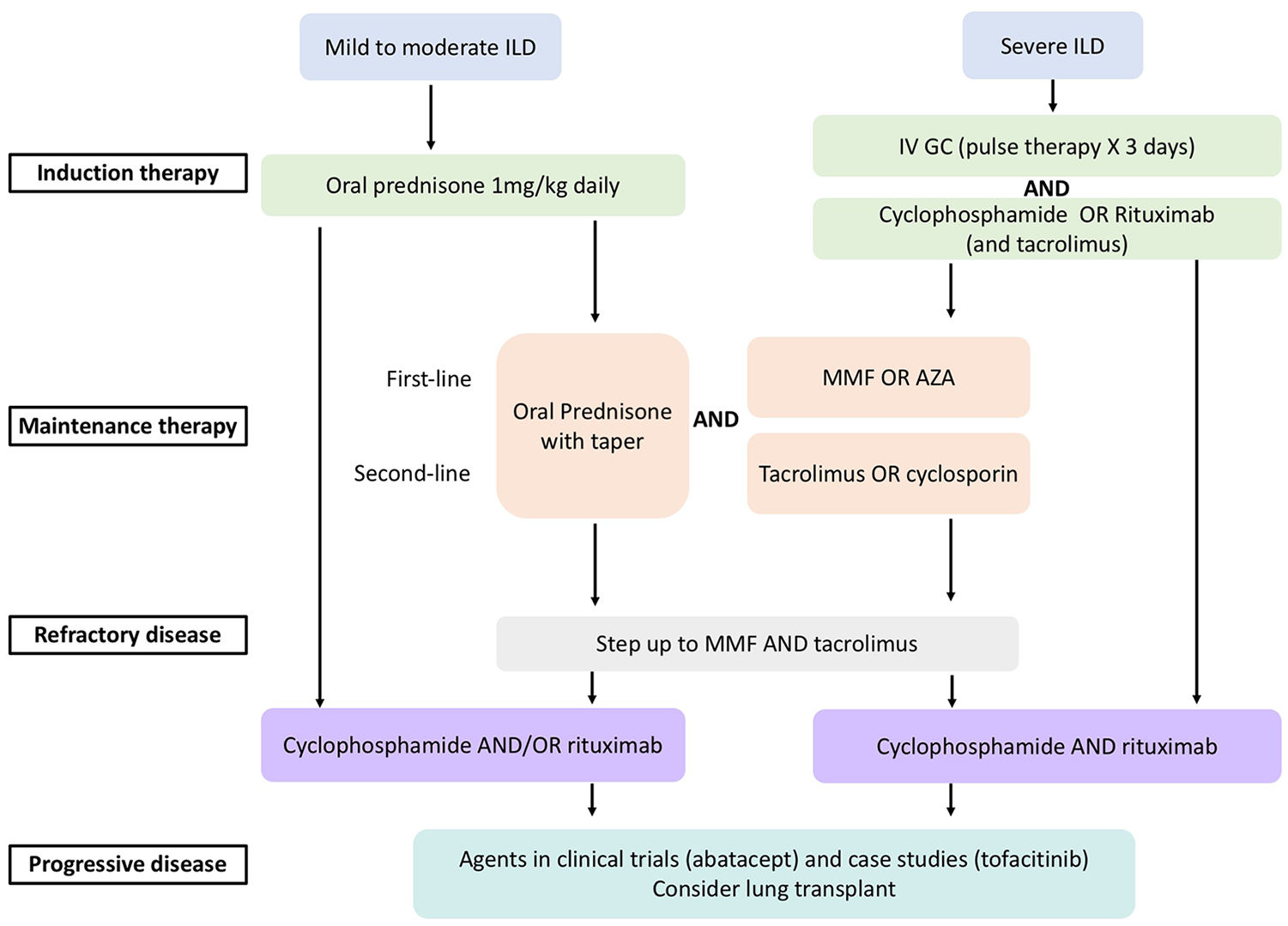
A proposed treatment algorithm for myositis-associated ILD (adapted from Oddis and Aggarwal 118 ).
Corticosteroids have long been first-line in the treatment of IIM but monotherapy with corticosteroid alone is rarely sufficient in ASSD, as ILD often recurs with corticosteroid tapering. Additional immunosuppressive agents are added for refractory skin, muscle, and/or lung disease as corticosteroid-sparing agents. There is no consensus or guidelines for additional immunosuppressive drugs, but frequently used agents include azathioprine, mycophenolate mofetil (MMF), tacrolimus, RTX, and CYC, which are summarized in Table 4. Methotrexate is very effective for ASSD with arthritis and/or myositis, but clinicians generally avoid methotrexate if ILD is also a predominant symptom, primarily due to potential difficulty in ascertaining the cause of worsening ILD to disease versus methotrexate pneumonitis. There is no evidence of increased methotrexate pneumonitis in non-RA diseases. 119 Intravenous immunoglobulin (IVIG) is beneficial when there is esophageal involvement, refractory rash, pregnancy, active infection, and calcifications but evidence of its use in myositis-associated ILD is limited to case reports only.120,121
Steroid-sparing agents for myositis-associated ILD.
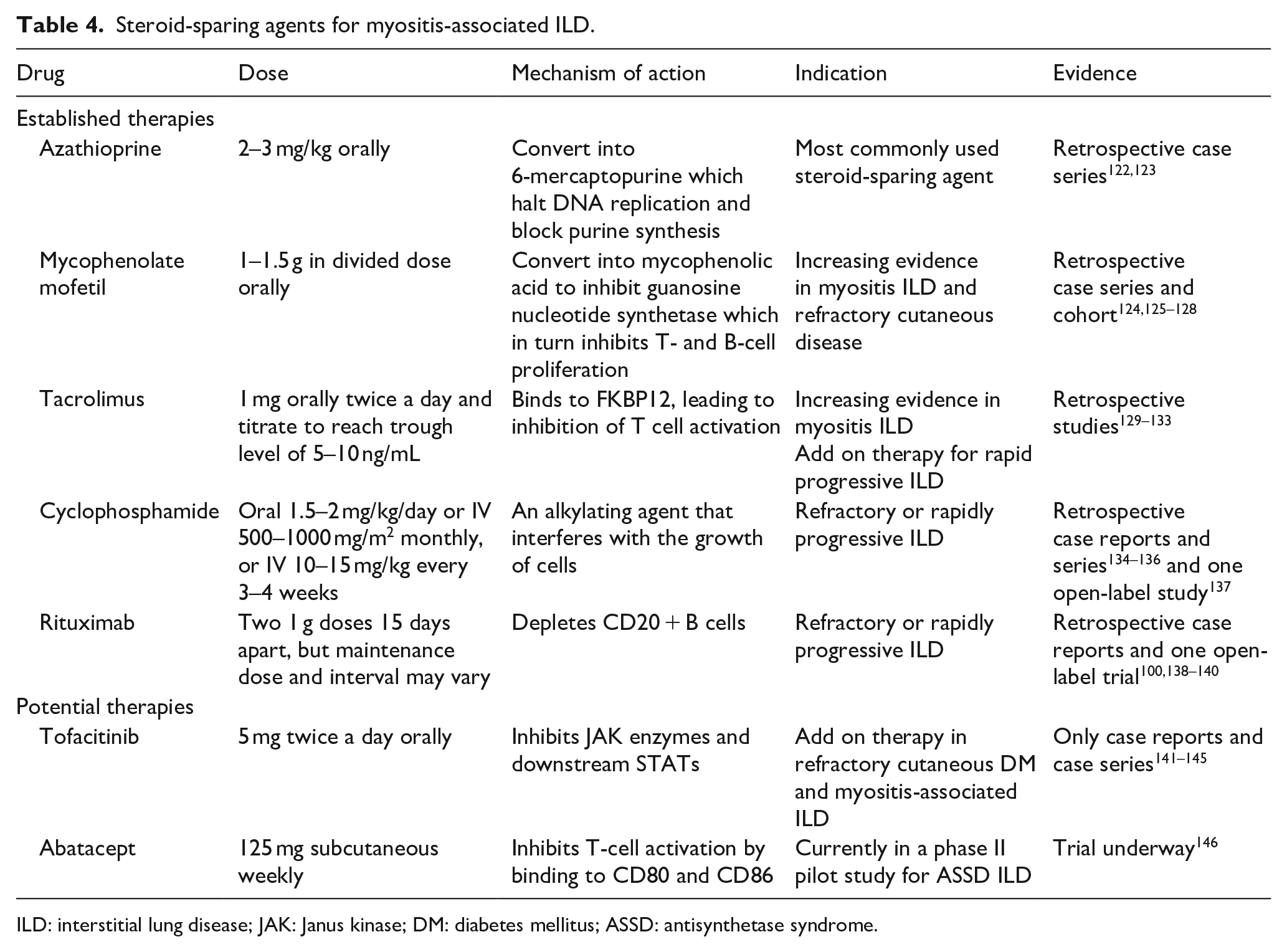
ILD: interstitial lung disease; JAK: Janus kinase; DM: diabetes mellitus; ASSD: antisynthetase syndrome.
Azathioprine treatment led to modest improvement in lung function in small retrospective case series or cohorts.122,123,147 Significant improvements in forced vital capacity (FVC) and DLCO were demonstrated in a large retrospective study of 125 patients with CTD-associated ILD (including 32 myositis patients) who were treated with MMF. 124 Benefit of CYC (IV or oral) in myositis-associated ILD has primarily been reported in retrospective case reports and case series.134–136 Clinically significant improvement was seen in dyspnea scale, oxygen requirement, FVC, and HRCT of the lung parenchyma. 136 In a prospective open-label study of 20 patients with myositis-associated ILD which included 10 RP-ILD, all 10 patients received IV CYC 10–12 mg/kg every 3 weeks for total 6–9 doses and resulted in stabilization, functional improvement, as well as increase in VC, FVC, and DLCO measures, with a median follow-up of 35 months. 137 Based on these evidences, we recommend IV CYC for severe and progressive myositis-associated ILD, in addition to the pulse glucocorticoid.
There is growing evidence of T-cell inhibition in the management of myositis-associated ILD. Cyclosporine and tacrolimus are the two available calcineurin inhibitors. For those with myositis-associated ILD, in whom cyclosporine was ineffective, small case series showed that tacrolimus treatment may improve lung disease.129–131 In vitro tacrolimus is 50-fold more potent than cyclosporine, 148 therefore tacrolimus serum trough levels should be monitored to ensure therapeutic level and to limit toxicity. In a retrospective review of 13 ASSD patients with ILD (12 anti-Jo-1 and 1 anti-PL-12) who were treated with tacrolimus for an average of 51 months, a significant improvement was observed in all pulmonary parameters measured as well as decline in creatine kinase (CK) and increase in muscle strength. 131 Calcineurin inhibitors have been shown to be effective in improving pulmonary function (PFT—pulmonary function test) and HRCT of chest 132 and should be used early 149 in progressive myositis-associated ILD. Asian studies have shown that addition of calcineurin inhibitor to conventional therapy led to longer event-free survival than treatment with conventional therapy alone.133,150,151
Increasing number of retrospective studies have shown benefit and safety of RTX in ILD associated with ASSD with reported objective improvement in PFT, ground-glass opacities and stability, or fibrosis on HRCT of chest.100,138,139,152–160 Those who had acute onset of ILD or had RTX within 12 months of disease onset had the best outcome. 139 In an open-label study of 10 patients with ASSD and refractory ILD, treatment with RTX resulted in a substantial steroid-sparing effect; clinically significant increase in FVC and/or DLCO was observed in 5 out of the 10 patients, stabilization in 4, and worsening in 1. 138 A retrospective study of 25 ASSD-ILD subjects treated with RTX from 2 US academic referral centers showed significant steroid-sparing effect, good tolerance, and improvement in CT imaging and/or PFT in most patients at 1- and 3-year follow-up. 159 In another retrospective study of 17 anti-Jo-1-positive ASSD patients who received RTX, 16 showed a more rapid and marked response than the others who were treated with conventional immunosuppressants (including CYC, cyclosporine, azathioprine, methotrexate, and leflunomide). This study also confirmed the effectiveness of RTX in those with refractory ILD, particularly in cases of concomitant high titers of anti-Ro 52. 100 Although not specific for ILD outcome, presence of ARS antibodies was predictive of better outcome in the post hoc analysis of the RTX in myositis trial. 161
In all of these studies, RTX is usually administered as two 1-g doses 2 weeks apart. Some supports to repeat RTX dosing over a single RTX cycle 159 but the subsequent dosing interval might vary, and no regimen has been standardized for the treatment of myositis with or without ILD. The RECITAL trial which is currently recruiting is the first randomized control trial to study the efficacy of RTX versus CYC as first-line treatment in CTD-associated ILD. 162
Tofacitinib has been shown beneficial in refractory cutaneous manifestation of DM.141,142 Its benefit as an additional therapy to CYC or RTX in refractory ILD was also demonstrated in recent case reports and case series.143–145 Abatacept is currently being assessed in a randomized controlled phase II trial to evaluate its efficacy, safety, and tolerability in ASSD associated ILD.
Treatment of other manifestations including myositis, arthritis, and cutaneous lesions parallels that of ILD summarized in Table 4. For refractory myositis or DM rash, IVIG can be effective. 163 For patients presented with predominant arthritis or cutaneous disease with no or minimal ILD, methotrexate or leflunomide can also be considered.
Summary
ASSD, although characterized by the classic triad of myositis, arthritis, and ILD, often presents with incomplete forms with only one or two of the manifestations in the presence of anti-ARS antibodies. Myositis in ASSD is clinically typical of PM (sometimes DM if rashes are present), but histopathologically distinct. Arthritis can be presenting symptoms and is often misdiagnosed as RA. ILD is the most common and often the presenting clinical feature in ASSD and is highly associated with higher mortality. Given that the anti-ARS antibodies are found in 6%–25% of idiopathic ILD, we consider testing anti-ARS in any ILD patients with or without other clinical features, as the presence of these antibodies confers different treatment and prognosis. PAH is common in ASSD; together with ILD, PAH confers a significant pulmonary mortality. We recommend doing screening echocardiogram at baseline for all ASSD patients and on follow-up for any evidence of exercise-induced desaturation or decline in DLCO on routine PFTs. For treatment of ASSD, conventional therapies include glucocorticoids usually in combination with one or multiple immunosuppressive drugs (DMARDs—disease-modifying antirheumatic drugs) like azathioprine, methotrexate, and MMF. Given the high prevalence of ILD, immunosuppressive agents that modulate T-cell function such as tacrolimus and deplete B-cells (RTX) are often required and helpful.
There is growing recognition that clinicoserological classification criteria for ASSD are needed. The new 2017 ACR/EULAR (European League Against Rheumatism) criteria do not capture the ASSD especially because the criteria weigh heavily on cutaneous and muscular findings which may be absent or evolve later in ASSD patients. 164 Presence of ILD or non-Jo-1 antibodies is not a part of the criteria. To overcome the shortcomings of 2017 EULAR/ACR myositis criteria and to promote clinical classification and early recognition of ASSD, currently there is a large multi-center international ACR/EULAR funded project for development and validation of ASSD classification criteria.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.