
Abstract
Background:
Hypogammaglobulinemia is a condition that can be related to both primary and secondary immunodeficiencies. While the role of primary immunodeficiency in immune-mediated diseases is well known, its occurrence in systemic sclerosis is not reported.
Objectives:
This study aims to describe the clinical features associated with hypogammaglobulinemia in a cohort of limited cutaneous systemic sclerosis patients.
Methods:
We retrospectively reviewed medical records of systemic sclerosis patients from two Italian referral centres (2010–2024). Included patients had limited cutaneous systemic sclerosis and presented reduced serum concentrations of one or more Ig isotypes (IgG < 700 mg/dL, IgA < 70 mg/dL or IgM < 50 mg/dL) in at least two separate measurements. Patients with secondary causes of hypogammaglobulinemia were excluded. Data collected included demographics, clinical features, Ig levels, infection history and comorbidities.
Results:
We identified 30 systemic sclerosis patients (93% female, mean age 62 years) with limited cutaneous involvement and hypogammaglobulinemia. Most patients were positive for anti-centromere antibodies and received periodic intravenous infusions of prostaglandin analogues. No patient received immunosuppressive therapy. Median (interquartile range) serum IgG levels 519.5 (175) mg/dL, median IgA 65.5 (48) mg/dL and median IgM 71.5 (49) mg/dL. Four patients who met the European Society for Immunodeficiencies (ESID) criteria for common variable immunodeficiency experienced recurrent infections and had associated immune-mediated diseases. Five patients had selective IgA deficiency, with frequent immune-mediated comorbidities (thyroiditis, Sjögren’s syndrome, arthritis, psoriasis). The other patients exhibited mild IgG deficiency without a significant infectious history.
Conclusions:
This is the first study describing a cohort of patients with limited cutaneous systemic sclerosis and hypogammaglobulinemia. Our population presented a high prevalence of immune-mediated comorbidities but low infection rates. Further research is needed to explore the underlying mechanisms and clinical significance of hypogammaglobulinemia in these patients.
Keywords
Introduction
Hypogammaglobulinemia is defined as the reduction of serum immunoglobulin (Ig) concentration. Serum IgG concentration below 500–600 mg/dL is considered the threshold for increased susceptibility to infections. 1 Antibodies are secreted by plasma cells, and they represent the main effectors of humoral immunity; their role is to recognize antigens and to trigger an inflammatory response capable of neutralizing pathogens. Usually, hypogammaglobulinemia has a bimodal distribution and is frequently diagnosed in the age groups between 6 and 10 years and in adults between 20 and 40 years, although there are increasing diagnoses in older patients.
Primary hypogammaglobulinemia is related to inborn errors of immunity. X-linked agammaglobulinemia, common variable immunodeficiency (CVID), selective/isolated Ig deficiency and hyper IgM syndrome represent the main primitive disorders of humoral system. However, low serum Ig levels can often be secondary to pathological conditions, such as haematological disorders, infections, neoplasms, nephrotic syndrome, enteropathies or linked to use of immunosuppressive agents. 1
The main clinical manifestations of hypogammaglobulinemia are recurrent infections, developmental disorders, immune-mediated diseases, lymphoproliferation, enteropathy and allergies, with higher incidence of neoplasms.
In clinical practice, secondary immunodeficiencies are often diagnosed in patients with immune-mediated diseases, such as systemic sclerosis (SSc), systemic lupus erythematosus (SLE), vasculitis and other conditions. Indeed, these patients are often subjected to immunosuppressive treatments, including rituximab (RTX), a monoclonal antibody directed against CD20 expressed by B lymphocytes,2,3 which has hypogammaglobulinemia as common adverse effect.
Interestingly, we have recently observed a peculiar subset of long-standing SSc patients with limited cutaneous involvement showing low serum Ig levels. SSc is a systemic immune-mediated disease characterized by Raynaud’s phenomenon, microangiopathy and fibrosis of skin, heart, lungs and gastrointestinal tract. 4 The most common SSc subset is limited cutaneous SSc (lcSSc), where skin involvement is restricted to face and the distal part of the four limbs and major organ involvement is less frequent than the diffuse type.5,6
The aim of this work is to describe the demographic and clinical characteristics of a series of lcSSc patients with hypogammaglobulinemia and elucidate its clinical significance.
Patients and methods
In this case series, we revised the medical records of patients with lcSSc followed up at two Italian referral centres (Clinica Medica, Marche University Hospital, Ancona and IRCCS Ospedale Policlinico San Martino, Genova, Italy) between January 2010 and January 2024. SSc diagnosis was made according to the American College of Rheumatology and European League Against Rheumatism (ACR/EULAR) classification criteria, 7 and lcSSc subset was defined according to LeRoy and Medsger. 8
Patients (⩾18-year-old) were included if they had an available quantitative immunoglobulin assay showing hypogammaglobulinemia of one or more Ig isotypes (IgA and/or IgG and/or IgM). Hypogammaglobulinemia was defined as a reduced serum Ig concentration compared to the reference values for the age group: we considered threshold values of IgG < 700 mg/dL, IgA < 70 mg/dL and IgM < 50 mg/dL. Low Ig levels had to be confirmed in at least two measurements more than 3 weeks apart in all patients. 9 CVID and selective IgA deficiency were diagnosed according to the European Society for Immunodeficiencies (ESID) criteria.10,11
Exclusion criteria were previous or current administration of immunosuppressive therapies (e.g. RTX, high-dose glucocorticoids (>7.5 mg/day), mycophenolate mofetil, cyclophosphamide, methotrexate, tocilizumab) and the presence of other causes that could explain hypogammaglobulinemia, including infections, current malignancy, nephrotic syndrome, nutritional disorders, severe enteropathy and other metabolic diseases.
Data collected for each patient included gender, age at SSc diagnosis, current age, disease manifestations, presence of autoantibodies, Ig levels, infectious recurrences, comorbidities and treatments. A diffusing capacity of the lungs for carbon monoxide (DLCO) below 75% was considered abnormal, while a pulmonary arterial systolic pressure (PASP) on echocardiogram ⩾36 mmHg was considered to be raised. Pulmonary arterial hypertension (PAH) was diagnosed by right heart catheterization (RHC) if the resting mean pulmonary arterial pressure (mPAP) was >25 mm Hg and the pulmonary arterial wedge pressure ⩽15 mm Hg. 12
All patients previously signed informed consent to data collection and publication. This study was conducted as part of standard clinical and laboratory practice and as such no specific approval was requested to Ethic Committee.
We presented data as frequencies and percentages for categorical variables, whereas continuous variables were expressed as mean and standard deviation or median (interquartile range (IQR)) as appropriate.
Results
Among 870 SSc patients from the two centres who had available data on serum Ig levels, we found 30 SSc patients that fulfilled the inclusion criteria and none of the exclusion criteria (Table 1–3). The mean age at SSc diagnosis was 45.6 years and the mean current age is 62 years; one patient died of macroangiopathic complications at the age of 88 years. Most patients were female (93%).
Demographic, clinical and laboratory characteristics of 30 lcSSc patients with hypogammaglobulinemia.
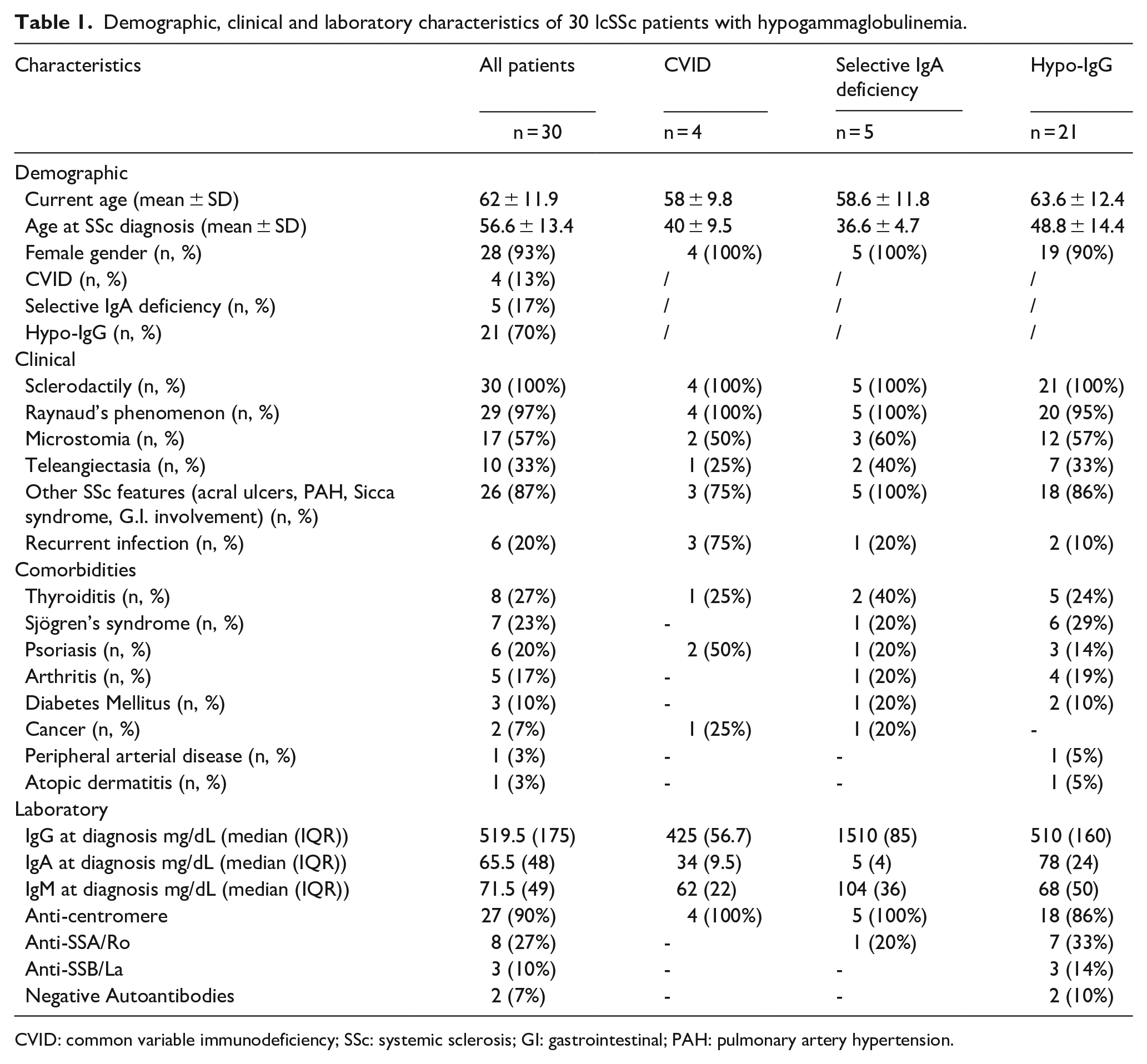
CVID: common variable immunodeficiency; SSc: systemic sclerosis; GI: gastrointestinal; PAH: pulmonary artery hypertension.
Baseline characteristics related to systemic sclerosis (SSc) of 30 lcSSc patients with hypogammaglobulinemia.
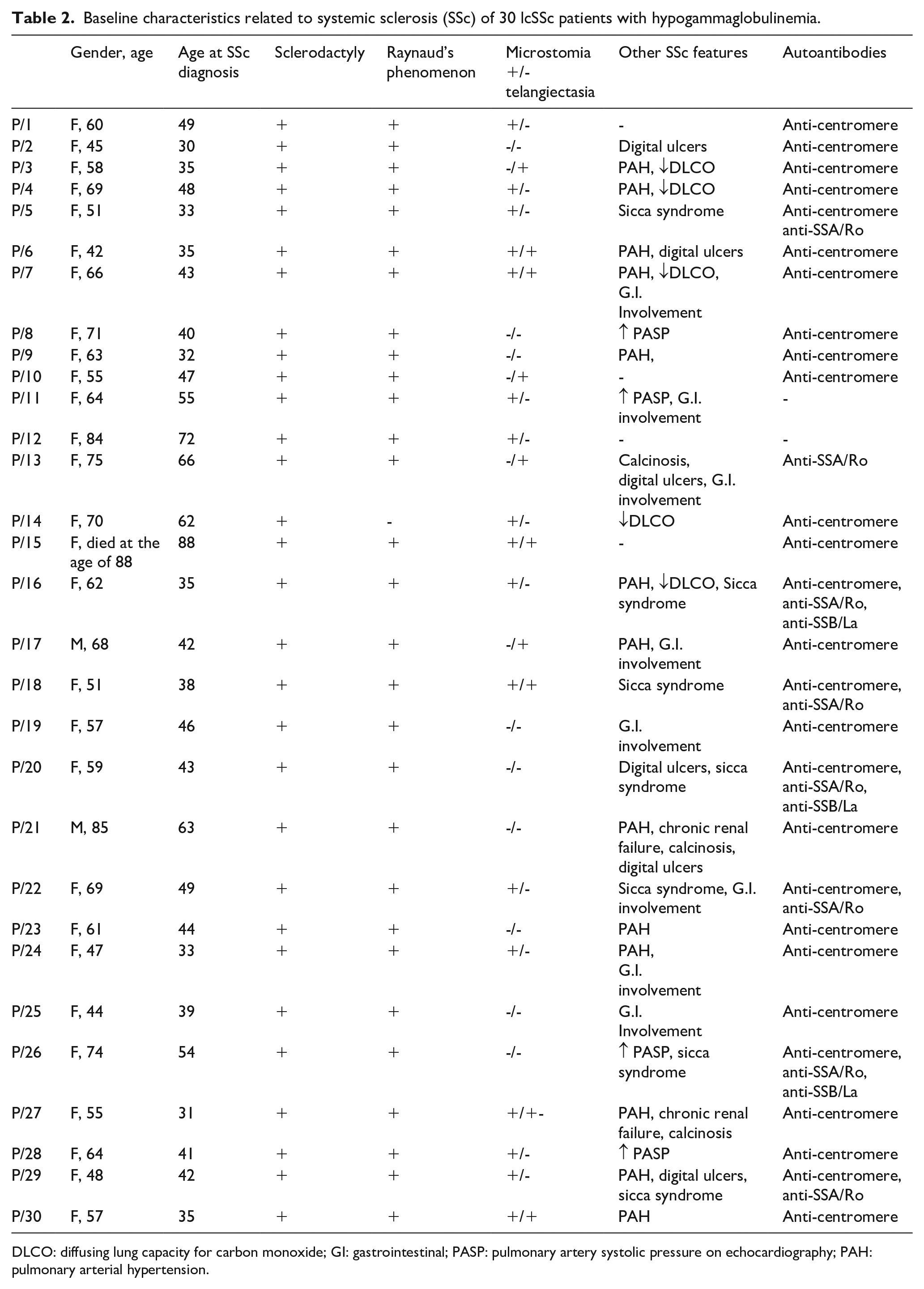
DLCO: diffusing lung capacity for carbon monoxide; GI: gastrointestinal; PASP: pulmonary artery systolic pressure on echocardiography; PAH: pulmonary arterial hypertension.
Main characteristics related to hypogammaglobulinemia of our patients with limited systemic sclerosis (SSc) and hypogammaglobulinemia.
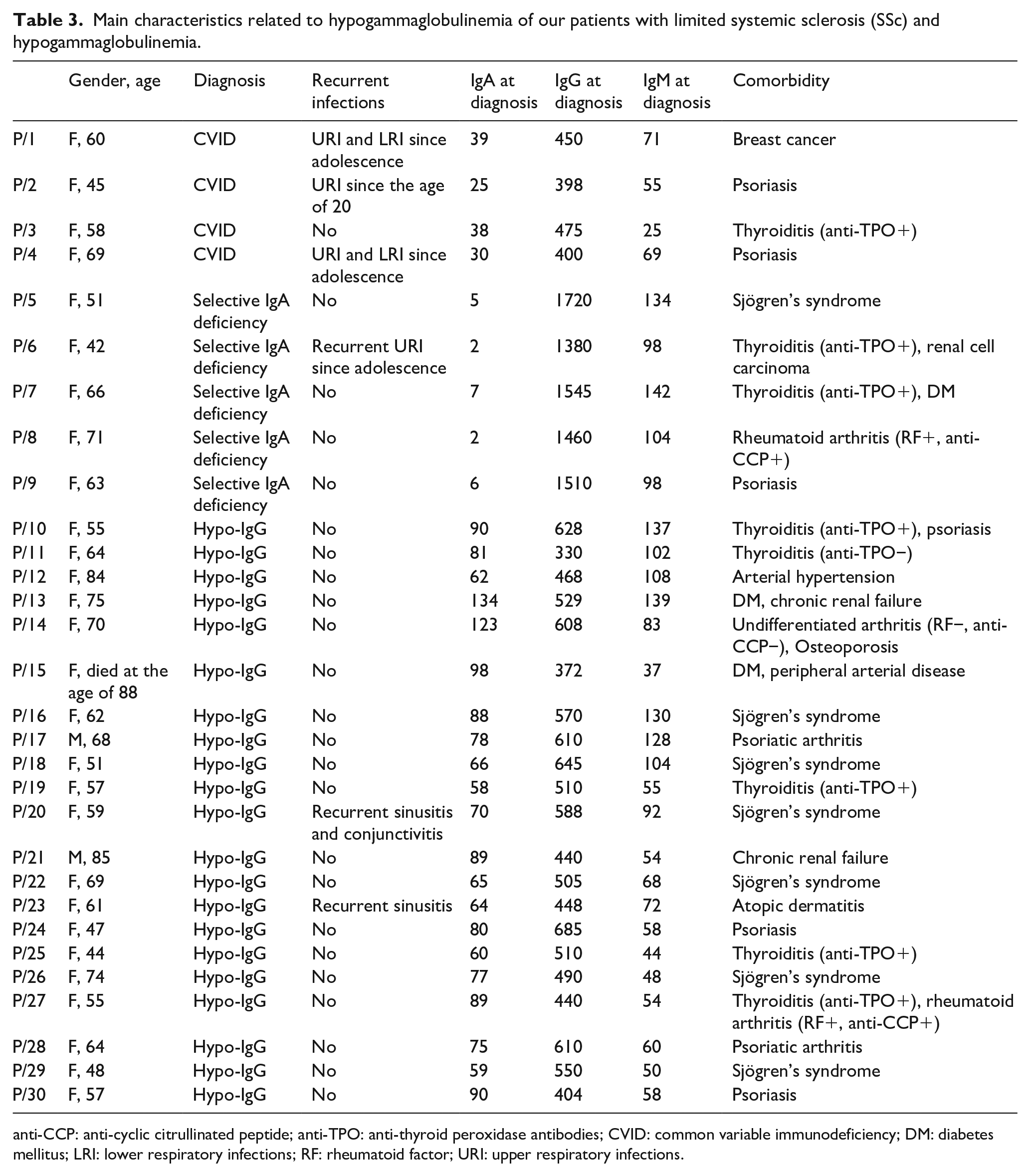
anti-CCP: anti-cyclic citrullinated peptide; anti-TPO: anti-thyroid peroxidase antibodies; CVID: common variable immunodeficiency; DM: diabetes mellitus; LRI: lower respiratory infections; RF: rheumatoid factor; URI: upper respiratory infections.
All the patients had lcSSc, characterized by Raynaud’s phenomenon, puffy fingers and skin’s thickening limited to the forearms, complicated in some cases by digital ulcers (20%) and PAH (43%). Other less frequent manifestations were sicca syndrome, microstomia, telangiectasia and gastrointestinal involvement (Tables 1 and 2). No patient had severe internal organ involvement, including interstitial lung disease (ILD). Most patients were positive to anti-centromere antibodies (ACA) (90%), and anti-SSA positivity was the second most common anti-nuclear antibody (ANA) specificity (Table 1). Median (IQR) serum IgG levels were 519.5 (175) mg/dL, IgA 65.5 (48) mg/dL and IgM 71.5 (49) mg/dL (Tables 1 and 2).
Most patients (75%) received periodic intravenous infusions of prostaglandin analogues for the treatment of Raynaud’s phenomenon. No patient received immunosuppressive therapy.
The most frequent comorbidities were other immune-mediated diseases, including psoriasis (n = 8), autoimmune thyroiditis (n = 8), Sjogren’s syndrome (n = 7) and arthritis (n = 5), including two cases of psoriatic arthritis and two cases of overlap rheumatoid arthritis (Table 3). Other cardiovascular and metabolic comorbidities, such as arterial hypertension, peripheral arterial disease, diabetes mellitus, chronic renal failure and osteoporosis, were also frequently observed (Table 3).
Among these cases, four female patients were diagnosed with CVID according to ESID criteria (P/1, P/2, P/3, P/4) (Table 3). Their mean current age was 58 years and three of them presented upper and lower respiratory tract infections from adolescence requiring frequent use of antibiotic therapy. In CVID patients, median (IQR) serum levels were IgG 425 (56.7) mg/dL, IgA 34 (9.5) mg/dL and IgM 62 (22) mg/dL. P/1 received CVID diagnosis 1 year after the SSc diagnosis and 20% subcutaneous Ig therapy was administered at the dose of 0.4 g/kg/month, with significant reduction in infectious events (currently no more than 1 infectious episode per year). She also developed breast cancer at the age of 42. The other three patients with SSc and CVID developed other immune-mediated comorbidities, P/2 and P/4 presented with psoriasis and P/3 had autoimmune thyroiditis.
Other five female patients presented selective IgA deficiency (IgA < 7 mg/dL) (P/5, P/6, P/7, P/8, P/9) (Table 3); mean current age was 59 years and only one of them reported bronchitis and upper respiratory tract infections from a young age. P/5 received selective IgA deficiency diagnosis 14 years after SSc; she also had an overlap with Sjögren’s syndrome characterized by severe sicca syndrome and anti-SSA/Ro positivity. Interestingly, her daughter (33-year-old) reported recurrent infections since adolescence with selective IgG1 and IgG3 deficiency. Both P/6 and P/7 developed thyroiditis, associated with renal cell carcinoma in the first patient and diabetes mellitus in the second one. Furthermore, P/8 presented with arthritis and P/9 with psoriasis.
Apart from these cases who received a well-defined diagnosis, the others showed a non-specific reduction in serum IgG levels, having median (IQR) IgG serum levels equal to 510 (160) mg/dL with normal IgG subclasses, IgA 78 (24) mg/dL and IgM 68 (50) mg/dL. Two of these patients reported recurrent upper respiratory tract infection (sinusitis) from adolescence (Table 3). As none of these patients reported recurrent infections, they did not undergo Ig replacement therapy.
Discussion
SSc is a rare and heterogeneous immune-mediated disease; the prevalence is estimated to range from 7.2 to 33.9 per 100,000 individuals and the annual incidence varies from 0.6 to 2.3 per 100,000 individuals in European Countries. The age at diagnosis is usually between 33.5 and 59.8 years with a higher prevalence in women, and a female:male ratio is equal to 3.8–11.5:1 in Europe. 13
In this case series, lcSSc patients with hypogammaglobulinemia were mostly female with a mean age at SSc diagnosis of 45 years. All patients presented a phenotype characterized by lcSSc involvement with a predominance of vasculopathy and sclerodactyly, without severe fibrotic manifestations. As such, they did not receive immunosuppressive therapies or have other conditions that could explain hypogammaglobulinemia. 14
The finding of hypogammaglobulinemia in SSc has been reported by very few studies if we exclude reduction in Ig levels secondary to immunosuppressive agents.2,3
In our series, four female patients received the diagnosis of CVID, based on the recurrent infections, reduced serum levels of IgG and IgA and/or IgM and impaired response to vaccination against tetanus. One patient (P/1) received the diagnosis of CVID at the age of 50, one year after SSc diagnosis. Indeed, the history of recurrent respiratory and urinary tract infections since childhood and the development of breast cancer 8 years earlier should have raised the suspicion of a possible immune deficit. Since she was administered Ig replacement therapy with 20% subcutaneous Ig therapy, the patient experienced a significant reduction in infectious recurrences and stability of vascular and cutaneous involvement of SSc. Previous reports documented a benefit of high-dose intravenous Ig (IVIg) with immunomdulatory role in SSc patients.15–17 The other three CVID patients all presented with immune-mediated diseases, which are the most frequent complications in CVID patients.
Balci et al. described a single case of a 53-year-old female patient affected by limited SSc who suffered from severe pulmonary infections which required hospitalizations, recurrent sinusitis and persistent low levels of Ig, associated with high-resolution computed tomography finding of bronchiectasis, so she was diagnosed with CVID and started IVIg (at 1 g/kg monthly), with clinical benefit. 18
Pamuk et al. reported another case about a 15-year-old male patient with X-linked agammaglobulinemia (XLA), that is a rare hereditary primary antibody immunodeficiency caused by mutations in the gene encoding Bruton’s tyrosine kinase (Btk) protein. Despite replacement therapy with IVIg, during follow-up he developed bronchiectasis, oligoarthritis, Raynaud’s phenomenon and thickening of the skin; laboratory tests revealed ANA positivity with a centromere pattern, and he received the diagnosis of limited SSc. 19
Five SSc female patients from our series had a selective IgA deficiency (SIgAD). SIgAD is a common primary antibody immunodeficiency characterized by an isolated reduction in IgA levels (< 0.07 g/L), which can be associated with mucosal infections, allergies and atopy, and autoimmune complications. Between 25.5% and 31.7% of SIgAD patients presented autoimmune organ-specific diseases with several proposed pathogenetic mechanisms, including predisposing HLA genes, monogenic mutation, lymphocyte and cytokine abnormalities, molecular mimicry with pathogens and dysregulation of immune pathways that protect against autoimmunity. In our series, we found that all SIgAD patients developed immune-mediated diseases, in particular thyroiditis (n = 2), psoriasis (n = 1), arthritis (n = 1) and Sjögren’s syndrome (n = 1). To emphasize the common genetic substrate that leads to the different immune-mediated manifestations seen above, the daughter of SIgAD patient P/5 presented a selective IgG1 and IgG3 deficiency with recurrent infections since adolescence.
Except for the patients reported above, the other patients had mild hypogammaglobulinemia that did not meet criteria for CVID or SIgAD diagnosis and were considered as having IgG deficiency (IgGD). 10 Ameratunga et al. 20 recently proposed a new classification for patients with hypogammaglobulinemia that does not meet CVID diagnostic criteria, dividing them into possible CVID (serum IgG ⩽ 500 mg/dL) or hypogammaglobulinemia of uncertain significance (HGUS with serum IgG 500–690 mg/dL). 20 Since these patients did not present a high rate of infectious recurrences, Ig replacement therapy was not indicated.
To the best of our knowledge, this is the first report of a possible association between lcSSc and low serum Ig levels, although the mechanisms underlying Ig reduction in these patients are unclear. A possible link may be that in this case series we observed a high risk of developing other immune-mediated diseases, both systemic and organ-specific. In fact, most patients had an associated immune-mediated disease, such as arthritis, psoriasis, thyroiditis and Sjogren’s syndrome. Thus, hypogammaglobulinemia in these cohorts may be the pre-existing condition leading to SSc. The susceptibility to autoimmunity in subjects with antibody deficiency may be regarded to as to a biological paradox. Although the mechanisms are not completely understood, there are some hypotheses on the possible co-existence of hypo- and hyper-immune states in the same patients, such as T and B lymphocytes abnormalities and/or the presence of autoreactive B cells and reduced T regulatory cells activity.21,22 These studies demonstrated the expansion of CD21low/−B cells and the reduction of T regulatory cells, switched memory B cells, naïve CD8+ and CD4+ T cells in the peripheral blood of patients with hypogammaglobulinemia. Furthermore, the increase in T helper 1 and follicular T CD4+ cells and thus in Th1 and interferon (IFN) gamma signalling has been described.21,22 Recent studies focused the attention on lymph nodes germinal centres and on human FOXP3+ T follicular regulatory cells (Tfr), a T regulatory subset that promote the synthesis of antibodies while controlling an exaggerated pathogenic autoreactive response. 23 An impairment in humoral response is responsible for antibody immunodeficiency or inadequate vaccine responses. On the opposite, the surge of autoreactivity could lead to immune-mediated disorders characterized by excessive autoantibody production, such as SLE and SSc, or by the presence of specific functional autoantibodies (such as anti-acetylcholine receptor antibodies in Myasthenia gravis).
Different T cell subtypes are deeply involved in the pathogenic process leading to fibrosis in SSc. They comprise T follicular helper (Tfh) cells, regulatory T cells (Treg), interleukin-17 (IL-17)-producing Th17 cells, CD4+ cytotoxic T lymphocytes (CTLs) and angiogenic T (Tang) cells with Th1/Th2 imbalance of Th1/Th2 and Th17/Treg. 24 Ricard et al. 25 investigated the population of circulating T follicular helper (cTfh) cells and their interaction with B cells in SSc. Besides an increased number of cTfh in patients with SSc compared with healthy controls, the authors documented an activated Tfh phenotype, mainly characterized by enhanced production of IL-21 in comparison with healthy controls. Finally, IL-21 has been demonstrated to be an effector of IFN-γ + IL-17 + Th17 cells in fibrosis process in SSc. 26
In the pathogenesis of SSc, both innate and adaptive immune cells are involved, with predominant Th2 responses, as highlighted by high levels of interleukin (IL)-13 and transforming growth factor-beta (TGF-β) in pathological tissues. Chronic inflammation and fibrosis are also sustained by the activity of B cells activated by IL-4 and IL-5 (Th2-derived cytokines) and IL-6, that stimulate the production of autoantibodies. All these pathways lead to the over-production of extracellular matrix by activated endothelial cells and fibroblasts, causing vasculopathy and unrestrained fibrosis of the skin and internal organs.27,28
We are aware of the limitations of this study, including the small sample size, which preclude any mechanistic insight. Extending the immunoglobulin profile to a larger cohort of SSc patients would help clarify the association, if any, between hypogammaglobulinemia and lcSSc.
Conclusion
In this case series, we described a peculiar phenotype of subjects with lcSSc and hypogammaglobulinemia, suggesting a possible link between the two conditions. Patients with SSc and hypogammaglobulinemia have a high rate of comorbid immune-mediated diseases, but most patients do not have infectious recurrences. The clinical significance of this observation and the involved mechanisms should be evaluated in further studies.
Footnotes
Acknowledgements
The authors thank all the patients who kindly agreed to participate in this study.
Authors’ note
The Editor/Editorial Board Member of JSRD is an author of this paper; therefore, the peer review process was managed by alternative members of the Board and the submitting Editor/Board member had no involvement in the decision-making process.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.