
Abstract
Electrolyte abnormalities are frequent in cancer patients, arising from the malignancy itself, anticancer therapies, or treatment-related complications. Hyponatremia, hypokalemia, hypomagnesemia, hypophosphatemia, and hypercalcemia occur through diverse mechanisms including endocrine dysfunction, renal tubular injury, gastrointestinal loss, cytokine-driven effects, and metabolic shifts during hematologic recovery. Novel agents such as immune checkpoint inhibitors, CAR-T therapy, targeted therapies, and transplant regimens have expanded both the spectrum and complexity of these disorders. These abnormalities can be clinically subtle yet contribute to morbidity, treatment delays, and poorer outcomes. Management requires timely recognition, targeted laboratory evaluation, and etiology-specific interventions such as hormone replacement, cytokine blockade, electrolyte repletion, and antiresorptive therapy. This review synthesizes current knowledge on therapy-associated electrolyte disturbances in oncology, emphasizing underlying mechanisms, clinical impact, and practical strategies for diagnosis and management.
Introduction
Electrolyte abnormalities, such as hyponatremia, hypokalemia, hypomagnesemia, hypercalcemia, and hypophosphatemia, are common in patients with cancer (Figure 1) and may result from a variety of mechanisms, including the malignancy itself, anti-cancer therapies, nutritional deficiencies, and organ dysfunction (Table 1). The primary aim of this review is to discuss the most common electrolyte disorders that arise as a direct consequence of cancer and cancer therapies. By highlighting these therapy-induced electrolyte derangements, the review seeks to enhance understanding of their clinical implications and management considerations.
Common electrolyte disorders associated with cancer.
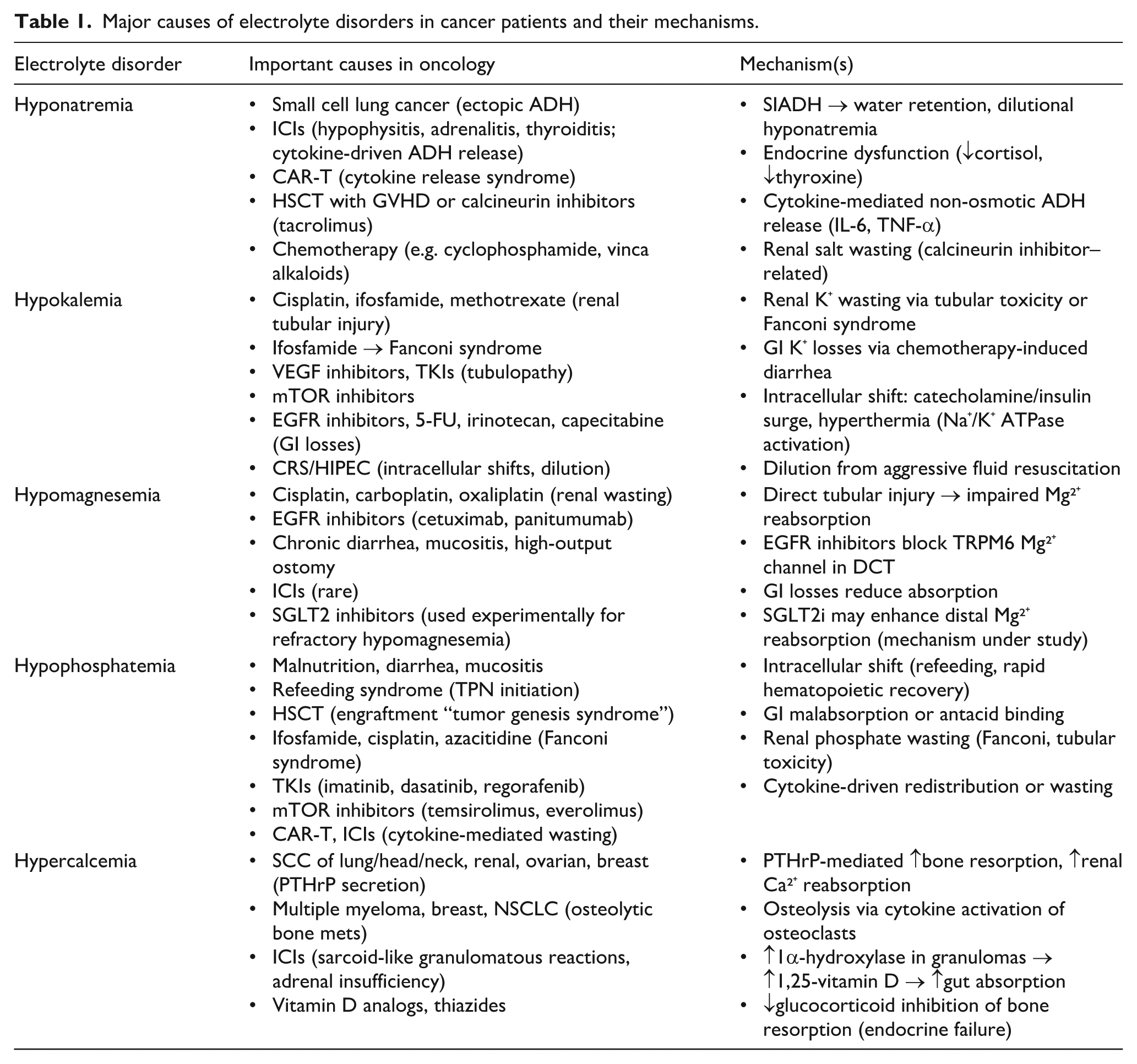
Major causes of electrolyte disorders in cancer patients and their mechanisms.
Hyponatremia
Case 1: A 66-year-old man with extensive-stage small cell lung cancer presents with progressive confusion and generalized weakness. He recently completed his first cycle of carboplatin, etoposide, and atezolizumab. On examination, he is euvolemic with stable vitals. Labs reveal a serum sodium of 119 mEq/L, serum osmolality of 255 mOsm/kg, urine sodium of 68 mEq/L, and urine osmolality of 620 mOsm/kg. TSH is 1.1 µIU/mL and morning cortisol is 14 µg/dL.
Hyponatremia is the most frequently encountered electrolyte abnormality in hospitalized patients with cancer and is associated with increased morbidity, treatment delays, and poorer overall health outcomes. 1 In oncologic populations, hyponatremia often arises from a complex interplay of malignancy-related factors, cancer-directed therapies, and treatment-related complications. The most common mechanism is the syndrome of inappropriate antidiuretic hormone secretion (SIADH), particularly in patients with small cell lung cancer (SCLC), where ectopic ADH secretion or hypothalamic stimulation via cytokines leads to water retention and dilutional hyponatremia. 2 Drug-induced SIADH is also common in cancer patients, particularly with the advent of immune checkpoint inhibitors (ICIs). ICI have introduced new mechanisms of hyponatremia through immune-related endocrinopathies and cytokine-mediated, non-osmotic ADH release. ICI therapy targeting PD-1, PD-L1, and CTLA-4 can result in autoimmune hypophysitis, adrenalitis, and thyroiditis, all of which may present with hyponatremia due to glucocorticoid or thyroid hormone deficiency. 3 Notably, anti-CTLA-4 agents such as ipilimumab appear to carry a higher risk of severe hyponatremia (serum sodium <124 mEq/L) compared to PD-1 or PD-L1 inhibitors, and combination ICI therapy is associated with increased risk of adrenal insufficiency (4%–9% vs 1%–2%), hypophysitis (13% vs 5%–8%), and thyroid dysfunction (20% vs 6%–10%) compared to single agent ICI therapy.3 –5 Beyond endocrinopathies, cytokine-driven, non-osmotic ADH release may contribute to acute hyponatremia in the setting of systemic inflammation induced by ICI therapy. Interleukin-6 (IL-6) is a central driver for non-osmotic vasopressin release in ICI-induced, inflammatory states. Real-world studies suggest that the incidence of hyponatremia in patients receiving ICIs is significantly underestimated in clinical trials. In a cohort of approximately 2500 patients treated with ICIs, 62% developed hyponatremia within 1 year, compared to an incidence of 9% in pooled randomized controlled trial data. 6
Chimeric antigen receptor T-cell (CAR-T) therapy represents another emerging cause of hyponatremia in cancer patients, primarily via cytokine release syndrome (CRS). The mechanism is like that of ICI-induced, non-osmotic ADH release mediated through pro-inflammatory cytokines. Most notably, IL-6, IL-1, TNF-α, and IFN-γ stimulate magnocellular neurons in the hypothalamus leading to water retention and dilutional hyponatremia. 7 One study exploring patients with diffuse large B-cell lymphoma receiving CAR-T reported a 15% incidence of serum sodium <130 mEq/L, with a strong inverse correlation between IL-6 levels and sodium concentration: approximately 2.7 mEq/L decrease per 10-fold increase in IL-6.7,8 Hyponatremia is also increasingly recognized in the hematopoietic stem cell transplant (HSCT) population. While not as common as other electrolyte disturbances, SIADH can occur in the context of graft-versus-host disease (GVHD), cord blood transplantation, or HLA-mismatched donor-recipient pairs. IL-6 and TNF-α, elevated post-transplant, are implicated in stimulating ADH release. 9 Additionally, calcineurin inhibitors such as tacrolimus, frequently used in GVHD prophylaxis, may contribute to hyponatremia by potentiating renal salt wasting or inducing SIADH-like effects through enhanced AVP sensitivity in the collecting duct. Corticosteroids, though less commonly associated with direct effects on sodium homeostasis, may also mask underlying adrenal insufficiency or contribute to volume changes that complicate the assessment of hyponatremia etiology.9,10
Evaluation of hyponatremia in cancer patients requires a methodical approach, incorporating serum and urine osmolality, urine sodium, and assessment of volume status. In cases where an endocrine cause is suspected, morning cortisol, ACTH, TSH, and free T4 levels should be obtained. Management depends on the underlying mechanism and severity of hyponatremia. For classic SIADH, fluid restriction, salt supplementation, and vasopressin receptor antagonists such as tolvaptan are the mainstays of therapy. In patients with endocrine dysfunction, hormone replacement with hydrocortisone or levothyroxine is essential. For those developing hyponatremia in the setting of CRS, targeted cytokine blockade with tocilizumab and systemic steroids is indicated. 8
Hypokalemia
Case 2: A 58-year-old woman with peritoneal carcinomatosis from appendiceal adenocarcinoma undergoes cytoreductive surgery followed by hyperthermic intraperitoneal chemotherapy (HIPEC) with mitomycin C and cisplatin. On postoperative day 2, she develops generalized weakness, diffuse abdominal pain and ileus. Labs show a potassium level of 2.9 mEq/L, magnesium 1.4 mg/dL, and a urine potassium of 42 mEq/L despite intravenous replacement.
Hypokalemia is a multifactorial electrolyte disturbance in patients with cancer that principally results from gastrointestinal loss, renal tubular dysfunction, intracellular shifts, or dilutional effects from aggressive fluid resuscitation. The clinical implications of persistent hypokalemia are substantial and can predispose patients to arrhythmias, neuromuscular weakness, and ileus. One of the most well cited causes of renal potassium loss is chemotherapy-induced tubular injury, particularly from agents such as cisplatin, ifosfamide, and methotrexate.11 –13 Cisplatin is particularly nephrotoxic; it can cause direct damage to the distal nephron, inducing a renal salt-wasting syndrome that includes hypokalemia and often concurrent hypomagnesemia, which can further impair renal potassium handling. 11 A case series of patients receiving cisplatin identified renal salt wasting as a rare but prolonged complication, persisting for up to 90 days following cessation of therapy in most patients, and even longer in isolated cases. 14 Ifosfamide is another alkylating agent that can induce Fanconi syndrome, a proximal tubulopathy characterized by urinary losses of potassium, phosphate, glucose, and bicarbonate. The risk is heightened when used in combination with cisplatin or at high cumulative doses. 13
Targeted therapies are also increasingly implicated in renal potassium wasting. Vascular endothelial growth factor (VEGF) inhibitors such as bevacizumab and tyrosine kinase inhibitors (TKIs) like sunitinib and axitinib have been linked to acquired Fanconi syndrome, likely due to direct tubular toxicity or microvascular injury. 15 Furthermore, mTOR pathway inhibition has been linked to renal potassium wasting, though the exact tubular mechanism remains incompletely defined. 16 Beyond nephrotoxicity, gastrointestinal losses are a major contributor to hypokalemia in oncology patients. Epidermal growth factor receptor (EGFR) inhibitors such as cetuximab, gefitinib, and erlotinib are well-documented causes, often inducing severe diarrhea and concurrent hypomagnesemia, both of which exacerbate potassium depletion. 17 Chemotherapy-induced diarrhea can lead to substantial potassium loss, particularly in regimens including 5-fluorouracil (5-FU), irinotecan, or capecitabine. 18
Hypokalemia is a frequently observed electrolyte disturbance in patients undergoing cytoreductive surgery followed by HIPEC arising from a convergence of renal, gastrointestinal, intracellular, and dilutional mechanisms. Renal potassium wasting is driven by the nephrotoxic potential of chemotherapeutic agents commonly used in HIPEC, including cisplatin, mitomycin C, and doxorubicin, with one study reporting postoperative hypokalemia and hypomagnesemia in up to 40% and 25% of patients, respectively. 19 Additionally, aggressive perioperative crystalloid resuscitation aimed at maintaining hemodynamic stability can increase urinary potassium losses through expansion of the extracellular fluid compartments and alteration of intravascular electrolyte concentrations. Recent intraoperative metabolic data show statistically significant reductions in serum potassium from approximately 3.57 ± 0.50 mEq/L pre-HIPEC to 3.28 ± 0.42 mEq/L post-HIPEC (p < 0.001). 20 Importantly, this study found that post-HIPEC potassium levels are an independent predictor of major postoperative complications. During the intraperitoneal hyperthermic phase, elevated intra-abdominal temperature stimulates Na⁺/K⁺-ATPase activity, promoting intracellular uptake of potassium. Concurrent catecholamine and insulin surges further shift potassium into cells, reducing extracellular levels. 21
Recognition of the underlying mechanism for hypokalemia is critical for effective management. Persistent hypokalemia despite adequate replacement should prompt evaluation for concurrent hypomagnesemia, which impairs renal potassium conservation. In select cases, such as chemotherapy-induced Fanconi syndrome or renal salt wasting, amiloride or potassium-sparing diuretics may reduce urinary potassium loss. This is generally accomplished by inhibitions of ENaC-mediated sodium reabsorption in the distal nephron and reducing downstream potassium secretion and kaliuresis.22,23 For patients with gastrointestinal loss, anti-diarrheal agents, mucosal protectants, and supportive care are first line. In all cases, serial monitoring and electrolyte repletion protocols should be integrated into routine oncologic care to avoid complications and maintain dose intensity of systemic therapies.
Hypomagnesemia
Case 3: A 63-year-old woman with metastatic colorectal cancer presents for routine follow-up during her eighth cycle of cetuximab-based chemotherapy. She reports mild fatigue and new-onset muscle cramps. Labs reveal a magnesium level of 0.8 mg/dL and potassium of 3.2 mEq/L. ECG shows mild QT prolongation. She has required intermittent intravenous magnesium infusions during prior cycles despite oral supplementation.
Hypomagnesemia is a frequently overlooked yet clinically significant electrolyte disturbance in patients with cancer. It can result from renal magnesium wasting, gastrointestinal losses, intracellular redistribution, or impaired dietary intake, mechanisms that are often exacerbated by oncologic therapies, including platinum agents, EGFR inhibitors, and immunotherapy. Cisplatin-based regimens cause renal magnesium wasting, leading to hypomagnesemia in up to 90% of patients, and concurrent hypokalemia may fail to correct unless magnesium stores are replenished first.24,25 Cisplatin is particularly notable for inducing sustained renal magnesium wasting due to damage to the distal convoluted tubule and thick ascending limb of Henle, where magnesium reabsorption primarily occurs. 24 Preclinical studies show that cisplatin accumulates in renal tissue at markedly higher concentrations than in plasma (e.g., ~5-fold) and remains elevated for a prolonged period post-dose, likely contributing to sustained nephrotoxicity. 26 The incidence of hypomagnesemia increases with cumulative exposure, affecting nearly all patients by the sixth cycle of cisplatin-based chemotherapy. 27 Furthermore, EGFR inhibitors impair renal magnesium reabsorption by inhibiting TRPM6, a magnesium channel expressed in the distal tubule. 28 In long-term users, especially those on cetuximab for metastatic colorectal cancer, the incidence of grade III/IV hypomagnesemia can approach 50%. 29
Other cytotoxic, platinum agents, including carboplatin and oxaliplatin, have also been associated with magnesium loss, though typically to a lesser extent. Additionally, gastrointestinal side effects of chemotherapy, such as diarrhea, mucositis, or high-output, ileostomy output can further deplete magnesium stores. These effects are particularly relevant in patients undergoing major debulking surgery and HIPEC. More recently, sodium-glucose cotransporter-2 inhibitors (SGLT2i) have shown promise in correcting refractory hypomagnesemia, especially in cisplatin induced hypomagnesemia. While their exact mechanism is still investigational, proposed explanations include increased vasopressin-mediated reabsorption and glucagon-induced activation of magnesium channels in the distal nephron. 30 In one case, a patient with long-standing, treatment-refractory hypomagnesemia achieved sustained normalization of serum magnesium levels following the initiation of dapagliflozin, an SGLT2i. 31
Management of hypomagnesemia in cancer patients typically relies on aggressive repletion strategies including both oral and intravenous supplementation, though high-dose oral formulations are often limited by gastrointestinal intolerance and should be avoided. Importantly, in patients with concurrent hypokalemia, magnesium should be corrected first to allow for renal potassium retention. For patients on EGFR inhibitors or with chronic GI losses, long-term maintenance therapy with scheduled infusions or innovative approaches like SGLT2i may be warranted.
Hypophosphatemia
Case 4: A 54-year-old man with relapsed acute myeloid leukemia undergoes allogeneic hematopoietic stem cell transplantation. On day +14 of engraftment, he develops new-onset confusion and proximal muscle weakness. Labs show a phosphorus level of 0.9 mg/dL, potassium 3.1 mEq/L, and magnesium 1.7 mg/dL. Renal function is normal. He has rising white blood cell counts and was recently started on total parenteral nutrition due to severe mucositis.
Hypophosphatemia is an under-recognized but clinically significant electrolyte disturbance in patients with cancer, particularly those experiencing intensive therapy or malnutrition. Phosphorus is critical for cellular energy metabolism, membrane structure, and red blood cell function. Severe deficiency (levels fall below 1.0 mg/dL (0.32 mmol/L)) can cause profound clinical consequences including hemolysis, respiratory muscle weakness, impaired leukocyte and platelet function, and seizures.32,33 Severe hypophosphatemia (<1 mg/dL) may result in rhabdomyolysis, cardiac dysfunction, and ventilator dependence in critically ill patients, and has been associated as an adverse prognostic marker in cancer cohorts. 34
The causes of hypophosphatemia in oncology are multifactorial. Decreased intake and malabsorption are common contributors, particularly in patients with advanced disease, gastrointestinal malignancies, or treatment-induced anorexia and diarrhea. Magnesium- and aluminum-based antacids, often used for mucositis or acid suppression, can bind phosphate in the gut and reduce absorption. 35 Transcellular phosphate shifts, as seen in refeeding syndrome, are particularly relevant in malnourished cancer patients who are initiated on nutritional support. In this setting, insulin release drives glucose and phosphate into cells, leading to rapid declines in serum phosphate levels. 36 Another relevant but less commonly described entity is tumor genesis syndrome, a phenomenon seen in aggressive hematologic malignancies (e.g. histiocytic lymphoma, Burkitt lymphoma, acute myelomonocytic leukemia) and post–allogeneic stem cell transplant. In this setting, rapid cell proliferation and hematopoietic recovery during engraftment sequesters phosphate intracellularly, mirroring the dynamics of refeeding syndrome. 37
Renal phosphate wasting is also a major contributor. Several chemotherapies are associated with acquired Fanconi syndrome, a proximal tubulopathy marked by phosphaturia, glycosuria, aminoaciduria, and bicarbonaturia. Ifosfamide, cisplatin, and azacitidine are common culprits. 38 Ifosfamide-induced Fanconi syndrome may persist even after therapy completion, particularly in young patients or those receiving high cumulative doses or concomitant nephrotoxic agents. Additionally, Fanconi syndrome may also occur in patients with monoclonal gammopathy, particularly due to the accumulation of filtered kappa light chains within the proximal tubules. This syndrome has been documented in adult T-cell leukemia/lymphoma where lymphomatous infiltration of the renal parenchyma impairs proximal tubule function.39,34
Hypophosphatemia is also increasingly recognized in association with novel therapies. TKIs such as imatinib, dasatinib, and regorafenib have been implicated, with incidences ranging from 2% to 7%. 41 mTOR inhibitors (e.g. temsirolimus, everolimus) also contribute through tubular toxicity, with reported incidences of hypophosphatemia as high as 22%. 34 Immune-based therapies, such as CAR-T therapy is associated with hypophosphatemia in over 50% of recipients, likely due to cytokine-mediated renal wasting or cellular redistribution during cytokine release syndrome. ICIs have also been implicated, with a pooled incidence of approximately 17%. 42
Hypercalcemia
Case 5: A 70-year-old man with newly diagnosed multiple myeloma presents with confusion, nausea, and constipation. Vitals are stable. Labs reveal a corrected calcium of 13.6 mg/dL, creatinine 1.8 mg/dL (baseline 1.0), phosphorus 2.2 mg/dL, and serum free light chains markedly elevated. ECG shows a shortened QT interval. He is not currently receiving any active therapy.
Hypercalcemia is a well-known complication of malignancy, occurring in approximately 10%–30% of patients with advanced cancer. It typically signals disease progression and carries a poor prognosis if not promptly recognized and treated. The pathophysiology of malignancy-associated hypercalcemia (MAH) generally involves one of four mechanisms: humoral secretion of parathyroid hormone-related peptide (PTHrP), osteolytic bone metastases, increased vitamin D activation, or treatment-associated disturbances in calcium homeostasis.
The most common mechanism of MAH is humoral hypercalcemia, which results from PTHrP secretion by tumor cells, particularly in squamous cell carcinomas (of the head and neck, lung, and esophagus), renal cell carcinoma, and ovarian and breast cancers. 43 PTHrP mimics the action of parathyroid hormone, increasing bone resorption and renal calcium reabsorption while decreasing phosphate reabsorption. Notably, serum PTH is typically suppressed in this setting. Osteolytic hypercalcemia is the second most common mechanism and is seen in cancers that preferentially metastasize to bone, such as multiple myeloma, breast cancer, and non-small cell lung cancer. Tumor cells in the bone microenvironment produce cytokines like IL-1, TNF-α, and prostaglandins that activate osteoclasts and lead to localized bone resorption. 44 Immune checkpoint inhibitors have introduced novel causes of hypercalcemia. Inflammatory sarcoid-like reactions induced by ICIs can lead to granulomatous hypercalcemia via increased expression of 1-alpha hydroxylase in activated macrophages, resulting in elevated 1,25-dihydroxyvitamin D and increased intestinal calcium absorption. 45 Other ICI-related mechanisms include adrenal insufficiency and ACTH deficiency, both of which reduce glucocorticoid-mediated inhibition of bone resorption and promote hypercalcemia. Treatment-related hypercalcemia can also occur, particularly with medications like vitamin D analogs or thiazide diuretics.
Initial management of hypercalcemia of malignancy depends on severity and symptomatology. For moderate to severe hypercalcemia (typically serum calcium >12 mg/dL), treatment includes aggressive intravenous hydration to promote calciuresis, calcitonin for short-term calcium reduction, and bisphosphonates (e.g. zoledronic acid or pamidronate) to inhibit osteoclast activity. In patients with refractory disease or poor renal function, denosumab, a monoclonal antibody targeting RANKL, may be preferred. Denosumab effectively reduces serum calcium by inhibiting osteoclast formation and activity. It has been particularly useful in bisphosphonate-resistant cases. 46 However, denosumab carries its own risks. Hypocalcemia is a well-recognized adverse effect, particularly in patients with advanced chronic kidney disease (CKD) or concurrent use of hypomagnesemia-inducing agents. Hypomagnesemia following denosumab use can further exacerbate calcium abnormalities and requires concurrent monitoring and correction. 47 Therefore, denosumab should be reserved for cases where bisphosphonates are ineffective or contraindicated due to renal dysfunction.
Conclusion
Electrolyte abnormalities are pervasive in cancer patients and reflect the complex interplay between malignancy, cancer-directed therapies, and host responses. As oncologic treatments continue to evolve—with the increasing use of immune checkpoint inhibitors, targeted therapies, CAR-T cell therapy, and intensive transplant regimens—the spectrum and mechanisms of electrolyte disturbances have expanded. Hyponatremia, hypokalemia, hypomagnesemia, hypophosphatemia, and hypercalcemia may arise through diverse pathways including endocrine dysfunction, renal tubular injury, gastrointestinal losses, cytokine-driven effects, and metabolic shifts during hematologic recovery.
Timely recognition and mechanistic understanding of these abnormalities are critical. Many of these disturbances can be subtle and nonspecific in presentation, yet they carry significant implications for treatment tolerability, morbidity, and patient outcomes. Incorporating routine electrolyte surveillance, maintaining a high index of suspicion in at-risk populations, and tailoring interventions to the underlying pathophysiology can help mitigate complications, reduce treatment delays, and improve overall quality of care in oncology.
Moving forward, greater awareness and interdisciplinary collaboration, particularly between oncologists and nephrologists, will be essential as the field continues to integrate novel therapies that affect electrolyte handling. Future research should aim to identify predictive biomarkers, optimize repletion strategies, and define the long-term consequences of persistent electrolyte imbalances in cancer patients.
Footnotes
Acknowledgements
The authors have no acknowledgments to disclose.
Data availability statement
Not applicable. No new data were generated or analyzed in the preparation of this manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable. This article is a narrative review and does not involve human subjects or animal research requiring ethical approval.
Informed consent
Not applicable. This is a review article and does not involve human participants or identifiable patient data.
Consent for publication
Not applicable.
Consent to participate
Not applicable.
Trial registration
Not applicable.
Grant number
Not applicable.
Guarantor
AR