
Abstract
Purpose:
To describe a case of retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S) to enhance early recognition of this often-missed diagnosis.
Methods:
A case report is presented.
Results:
A 50-year-old woman with a history of Raynaud phenomenon, memory difficulties, and a family history of strokes was referred for evaluation of a bilateral, small-vessel, occlusive disease refractory to immunosuppressive therapy. An extensive workup for treatable causes was unrevealing. Fifteen months after presentation, brain imaging showed white-matter lesions and dystrophic calcification, which led to the discovery of a pathogenic variant in TREX1 and the diagnosis of RVCL-S.
Conclusions:
Retina specialists play a critical role in the timely diagnosis of RVCL-S. Although the findings in this condition can mimic those in other common retinal vascular disorders, there are key characteristics that increase the suspicion for RVCL-S. Early recognition might decrease unnecessary therapies and procedures.
Keywords
Introduction
Retinal vasculopathy with cerebral leukoencephalopathy (RVCL-S) is a rare and devastating genetic disorder that primarily affects the small vessels of the retina and central nervous system (CNV). 1 The condition usually presents in the third or fourth decade of life with visual symptoms from the retinal vasculopathy followed by cerebral dysfunction associated with white-matter brain abnormalities seen on magnetic resonance imaging (MRI). 2 The correlation with neurologic manifestations might not be initially apparent because they do not present simultaneously.
The retinopathy mimics several disorders, including diabetic retinopathy (DR), ocular ischemic syndrome, hypercoagulable conditions, or inflammatory vasculitis. Misdiagnosis of the retinopathy could lead to ineffective treatment with immunosuppressive agents. The cerebral lesions might be consistent with multiple sclerosis or glioblastoma, which potentially leads to unnecessary brain biopsies or other invasive tests.
We present a case of progressive, symptomatic, noninfectious, occlusive RVCL-S that led to a diagnosis of retinal vasculopathy with cerebral leukoencephalopathy. Our objective was to heighten the awareness and earlier recognition of this rare but important disease.
Case Report
A 50-year-old woman presented with a 6-month history of decreased sharpness in the vision in both eyes. The ocular history was significant for a choroidal nevus in the left eye. The medical history was remarkable for Hashimoto thyroiditis, hypertension, hyperlipidemia, miscarriage, Raynaud phenomenon, and migraine headaches. She also had a recent history of fatigue with transient memory difficulties.
The family history was noteworthy for presumed cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) in her father and paternal uncle; both died in their 5th decade as a result. Her 54-year-old sister was recently diagnosed as having uveitis with intraocular hemorrhage and glaucoma.
At presentation, the patient’s visual acuity (VA) was 20/20 OU. The intraocular pressure, extraocular movements, pupil, and anterior segment examination were normal in both eyes. Funduscopic examination showed normal optic nerves with cotton-wool spots and intraretinal hemorrhages in the macula and the midperiphery in both eyes (Figure 1, A and B). There were sclerotic vessels in the inferior temporal periphery in both eyes and a 9500 µm faint flat area of poorly demarcated choroidal pigmentation temporal to the left macula. There was no evidence of vitritis or vitreous hemorrhage in either eye.
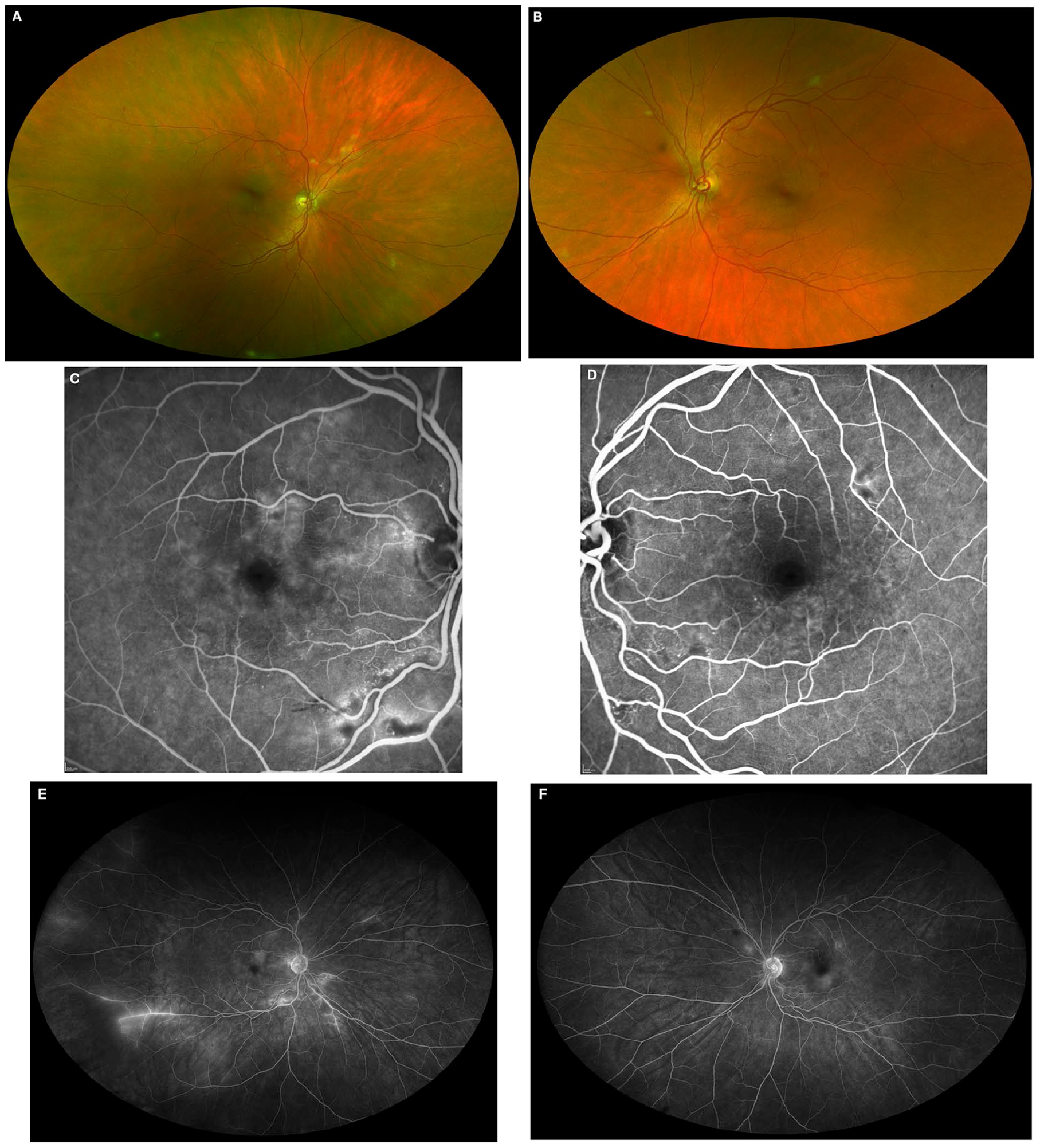
Ultra-widefield fundus images captured by confocal scanning laser ophthalmoscopy (A and B) show cotton-wool spots and intraretinal hemorrhages in the macula and midperiphery of both eyes. There are sclerotic vessels in the inferior temporal periphery of both eyes and a 9500 µm flat area of poorly demarcated choroidal pigmentation in the left temporal periphery. Midphase fluorescein angiography captured by 30-degree confocal scanning laser ophthalmoscopy (C and D) shows small capillary occlusions with leakage corresponding to the cotton-wool spots. Fluorescein angiography captured by ultra-widefield confocal scanning laser ophthalmoscopy (E and F) shows leakage at sites of capillary occlusion plus inferior temporal nonperfusion with adjacent vascular staining and leakage.
Fluorescein angiography (FA) showed several foci of capillary nonperfusion and telangiectasias with late leakage in the macula and midperiphery in both eyes (Figure 1, C and D). There was temporal ischemia with staining of the blunted vasculature in the right eye (Figure 1E). No peripheral ischemia was seen in the left eye (Figure 1F). The ocular coherence tomography (OCT) images showed focal hyperreflective thickening of the nerve fiber layer corresponding to the cotton-wool spots. There was no evidence of macular edema.
An extensive workup included evaluations for antineutrophil cytoplasmic antibody/antinuclear antibody–associated vasculitis, blood dyscrasia, diabetes mellitus, immunodeficiency, gammopathy, sarcoidosis, Bartonella, cytomegalovirus, hepatitis B and C, human immunodeficiency virus, human T-lymphotropic virus, Lyme disease, syphilis, toxoplasmosis, tuberculosis, West Nile virus, Whipple, CADASIL, and Susac syndrome. The findings were unremarkable. MRI of the brain showed mild parenchymal volume loss but no demyelinating lesions.
Fifteen months after initial presentation, the patient developed visual auras, dimmed vision, and a blind spot in the temporal portion of her left eye. The VA remained 20/20 OU. A dilated fundoscopic examination showed new cotton-wool spots and intraretinal hemorrhages in both eyes. OCT angiography showed focal areas of attenuated capillary flow in both eyes. There were also new areas of leakage on ultra-widefield FA in both eyes. Magnetic resonance angiography (MRA) of the brain and a computerized tomography (CT) angiogram of the brain and neck showed no evidence of vasospasm, arteriovenous malformation, flow-limiting stenosis, or branch occlusion.
Progression of the occlusive vasculopathy prompted initiation of treatment with mycophenolate mofetil and dexamethasone implants in both eyes. Mycophenolate mofetil alone was ineffective; therefore, adalimumab 40 mg was added. Despite 7 months of treatment with combined 500 mg mycophenolate mofetil daily and 40 mg adalimumab every other week, there was evolution of the deep capillary occlusions and peripheral vascular staining and leakage in both eyes. Because the patient was unable to tolerate higher doses of the current medications, azathioprine 25 mg daily was initiated.
Five days after azathioprine was introduced, the patient presented to the emergency department with vomiting and a frontal headache that had awoken her from sleep. Brain and spine imaging, consisting of an MRI, MRA, magnetic resonance venography, and CT scan, showed T2 hyperintensities in periventricular white matter, left frontal white matter, and left cerebellar hemispheric white matter (Figure 2). Dystrophic calcification was seen in the left frontal lobe. Cerebral spinal fluid testing was normal with no evidence of inflammatory or immune-mediated biomarkers. A 4-vessel cerebral angiogram was performed and did not show evidence of vasculitis affecting the large or medium-size arteries.
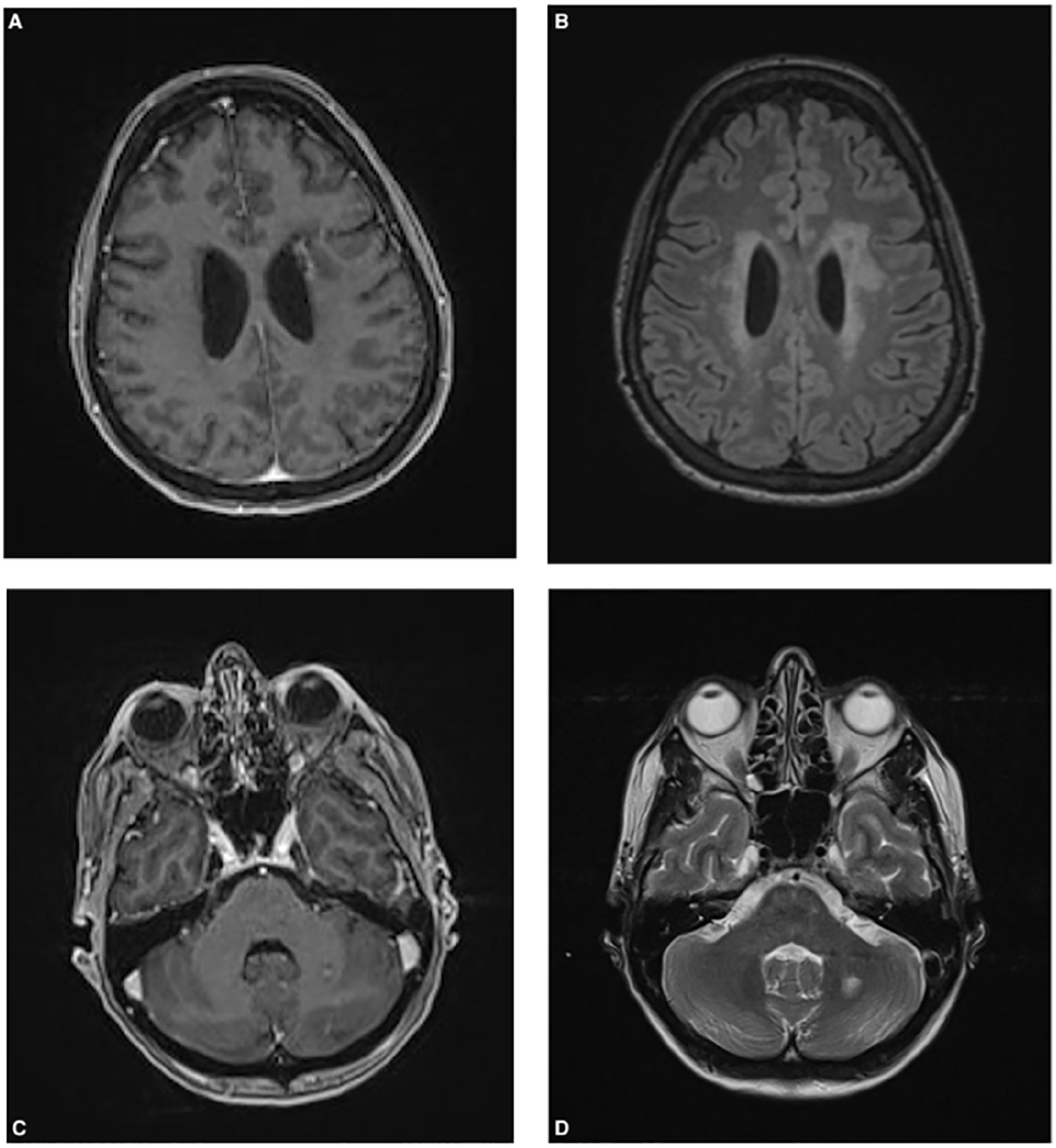
Magnetic resonance imaging of the brain shows confluent white matter T2-weighted fluid-attenuated inversion recovery hyperintensities around the lateral ventricles (B), with more focal left frontal and left deep cerebellar white-matter lesions (B and D). Dystrophic calcification with linear enhancement within left frontal periventricular lesion (A) and punctate enhancement in left deep cerebellar white-matter lesion (C).
VA remained 20/20 OU. A fundoscopic examination showed additional cotton-wool spots, telangiectasias, and intraretinal hemorrhages in both eyes (Figure 3, A and B). Enlarged areas of decreased deep capillary flow signal and widening of the foveal avascular zone were seen in both eyes on OCT angiography (Figure 3, C-F). Ultra-wide field FA showed new areas of leakage, progression of the peripheral ischemia, and staining of vasculature in both eyes (Figure 3, G and H).
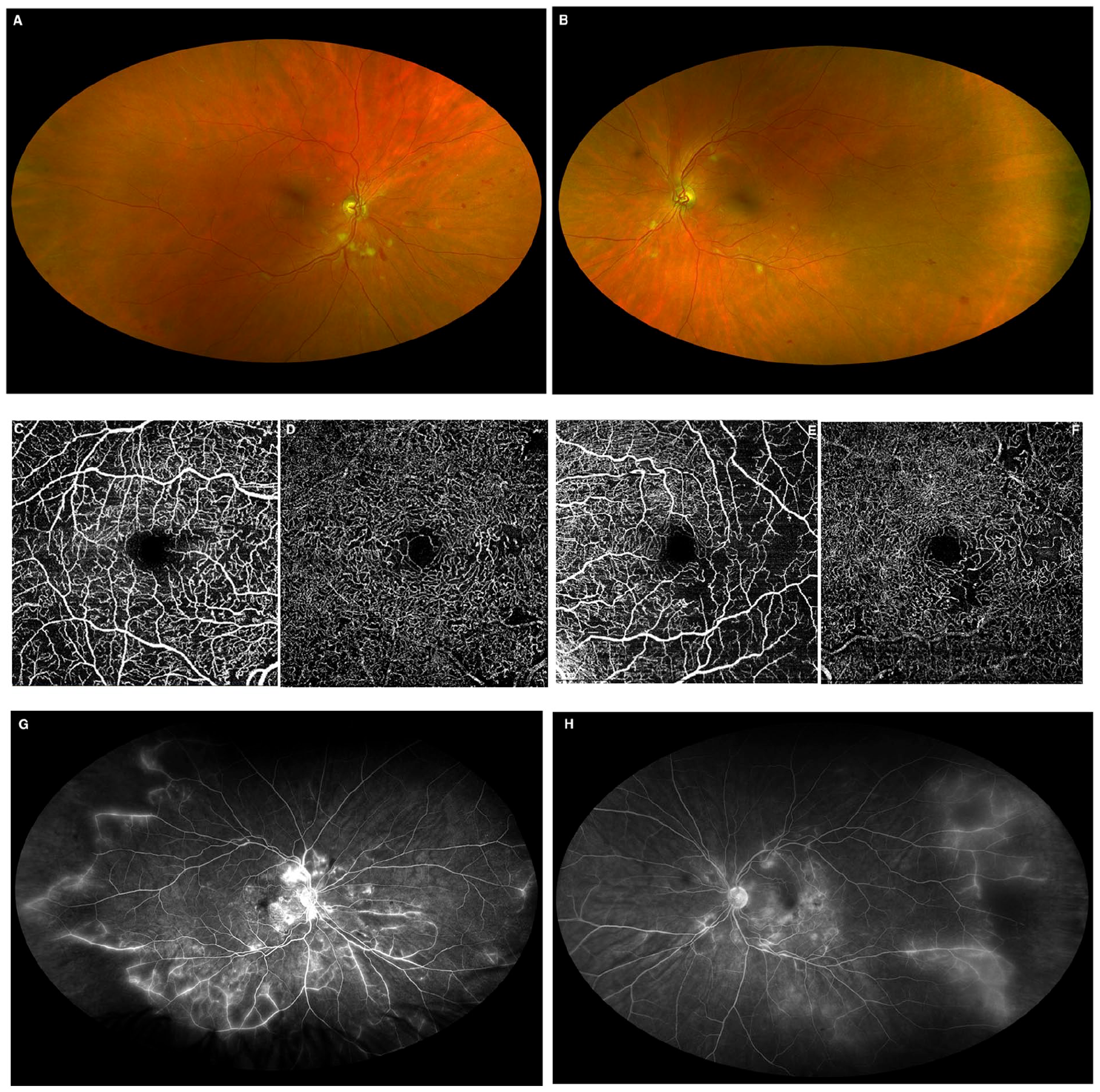
Ultra-widefield fundus images captured by confocal scanning laser (A and B) show new cotton-wool spots and additional intraretinal hemorrhages and telangiectasias in both eyes. There is persistent vascular sheathing in the inferior temporal periphery of the right eye and new vascular sheathing in the inferior temporal periphery of the left eye. Optical coherence tomography angiography (C-F) shows enlargement of the foveal avascular zone in both eyes with areas of decreased superficial (C and E) and a deep capillary flow signal (D and F). The fluorescein angiography captured by confocal scanning laser ophthalmoscopy (G and H) shows increased vascular staining and leakage with progression of the capillary dropout and vascular remodeling seen in Figure 1, E and F.
The neurological presentation raised suspicions for a hereditary cerebral vasculopathy. Subsequent genetic testing showed a heterozygous pathogenic variant in TREX1 [c.703dupG (p.V235GfsX6)], resulting in a diagnosis of RVCL-S. The immunosuppressive medications were discontinued, and the patient was referred for inclusion in a phase 2 clinical trial using crizanlizumab (clinicaltrials.gov identifier, NCT04611880).
Conclusions
RVCL-S is an autosomal-dominant, systemic, small-vessel endotheliopathy that affects highly vascularized tissues, including the retina, brain, liver, and kidneys. 1 Before recognition of a common C-terminal frameshift mutation in TREX1, RVCL-S was known as 3 distinct diagnoses; that is, cerebroretinal vasculopathy, hereditary endotheliopathy with retinopathy, and nephropathy and stroke with hereditary vascular retinopathy. 1
The mechanism by which this mutation affects the microvasculature has not been defined. TREX1 pathogenic variants that are associated with other conditions, such as systemic lupus erythematosus (SLE) and autosomal-dominant familial chilblain lupus, result from absent exonuclease activity. In RVCL-S, the exonuclease activity is preserved. Instead, the mutation removes the C-terminus, which anchors the protein to endoplasmic reticulum, thereby altering the intracellular location. 1 It has been postulated that the aberrant location of TREX1 in RVCL-S results in impaired repair of oxidative DNA damage, leading to detrimental accumulation of TREX1 in endothelial cells. 3 Histopathologic studies of the vasculature in patients with RVCL-S found abnormalities of the vascular basement membrane and hyalinization of the arterioles.1,3
As of this writing, RVCL-S has been identified in 44 families (email communication with Jonathan Miner, MD, PhD, May 2022). It is highly possible that RVCL-S is underdiagnosed as a result of unfamiliarity with the disorder, heterogeneous phenotypes, and asynchronous retina and systemic manifestations, which can mimic other disorders. Ophthalmologists play a critical role in the diagnosis of RVCL-S because the presenting feature is usually a progressive occlusive vasculopathy. 1
Grand et al 4 first characterized the ophthalmic features of RVCL-S using standard 30-degree FA. The findings included bilateral multifocal microvascular abnormalities of the posterior pole, including telangiectasia, microaneurysm, capillary dropout with eventual enlargement of the foveal avascular zone, and cotton-wool spots. Subsequent reports described evidence of peripheral retinal nonperfusion, which might eventually lead to retinal neovascularization and vitreous hemorrhage.5–8 More recently, structural OCT and OCT angiography studies have confirmed the location of infarctions to be at both the superficial and the deep capillary plexuses. 6 These findings correspond to the presence of cotton-wool spots and paracentral acute middle maculopathy lesions, respectively.
Our patient had an early stage of RVCL-S, with capillary obliteration, cotton-wool spots, vascular remodeling, and peripheral nonperfusion. The patient’s sister was subsequently diagnosed with RVCL-S. Although we did not evaluate the sister, we speculate that the intraocular hemorrhage and glaucoma likely represent the sequelae of neovascularization that can occur in more advanced retinal vasculopathy.
The retinal findings of RVCL-S are followed shortly by focal neurologic symptoms (hemiplegia, dysarthria, apraxia) and cognitive impairment with intracerebral mass lesions and white-matter abnormalities.1,5 Characteristic features of brain imaging include rim-enhancing mass lesions, hyperintense punctate images with nodular enhancement and possible calcifications, and multiple focal nonenhancing punctate hyperintense white-matter lesions with nodular enhancement. 1 Patients can also develop liver disease, nephropathy, and anemia with or without gastrointestinal bleeding. 1 The migraine headaches and Raynaud phenomenon described by our patient are common in RVCL-S and typically precede the onset of retinopathy. 1
Our patient’s diagnostic dilemma stemmed from the extensive differential diagnosis for retinal vasculopathy. In its earliest stages, RVCL-S may resemble many retinal vascular disorders, including DR, ocular ischemic syndrome, radiation retinopathy sickle cell retinopathy, phospholipid antibody syndrome, Susac syndrome, Eales disease, viral conditions, syphilis, and vascular conditions (eg, antineutrophil cytoplasmic autoantibody–associated vasculitis, Behçet disease, polyarteritis nodosa, SLE).
In the presence of CNV pathology, the differential remains broad and includes sarcoidosis, Susac syndrome, viral infection, Lyme disease, toxoplasmosis, tuberculosis, and syphilis as well as vascular conditions such as Behçet disease, granulomatosis with polyangiitis, polyarteritis nodosa, and SLE. Inherited neurodegenerative disorders that could produce similar brain MRI findings include cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy, cathepsin A–related arteriopathy with strokes and leukoencephalopathy, and cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy. 2 However these conditions typically do not result in visual symptoms.9–11
A distinguishing feature of early RVCL-S is the appearance of a refractory occlusive retinal vasculopathy coupled with a strong family history of cognitive problems, atypical vasculitis symptoms, and mortality during middle age. As with systemic vasculitides, such as granulomatosis with polyangiitis and SLE, RVCL-S is a small-vessel occlusive disease rather than a retinal vasculitis. Therefore, the characteristics of retinal vasculitis, including intraocular inflammation, vascular sheathing with perivascular exudates, and cystoid macular edema, are not typically found in RVCL-S. This helps differentiate RVCL-S from conditions such as Behçet disease and sarcoidosis. The later stages of RVCL-S involve cognitive decline and white-matter brain lesions.1,2 Although the brain abnormalities might be periventricular, as in multiple sclerosis, they are more confluent and lack the round or ovoid appearance as well as the perpendicular orientation to the ependymal surface typical of multiple sclerosis lesions.
For ophthalmologists to suspect a diagnosis of RVCL-S, it is critical to perform a dilated eye examination, FA, and OCT imaging with a comprehensive review of systems, medical history, and family history. The diagnosis of RVCL-S is confirmed molecularly by identifying a heterozygous pathogenic variant in TREX1.
The relentless neurological deterioration in RVCL-S eventually results in premature death.1,2 Although no available disease-modifying treatment is available for RVCL-S, recruitment for a phase I clinical trial (NCT02723448) investigating aclarubicin was recently completed. Aclarubicin is an anthracycline antibiotic that suppresses the endoplasmic reticulum glycan pathway. At present, a phase 2 clinical trial (NCT04611880) is active; it is investigating crizanlizumab for the reduction of ischemia and brain lesions in RVCL-S. Crizanlizumab is a humanized monoclonal anti-P-selectin antibody that prevents leukocyte adhesion to the vascular endothelium, thereby limiting risk of microvascular occlusion. 11
Immunosuppression has not been found to be a beneficial therapy for RVCL-S. 1 Therapy aimed at treating the complications of retinal ischemia, such as intravitreal injections of antivascular endothelial growth factor or photocoagulation, might improve vision. Corticosteroids have been found to reduce the cerebral vasogenic edema.1,7
Given the potential implications for family planning and the potential for early management of the retinal vasculopathy in relatives, genetic counseling plays a crucial role in the management of RVCL-S patients. At present, there is no consensus on the follow-up frequency for individuals with RVCL-S. At a minimum, annual ophthalmic evaluations, commencing in the 4th decade, have been suggested. 2 The frequency of these examinations would change as appropriate based on disease progression.
In conclusion, because the potential for misdiagnosis is great, it is possible that RVCL-S is an under-recognized condition. Retina physicians are usually the first specialists these patients present to, and they play an essential in the accurate and early diagnosis of RVCL-S. A diagnosis of RVCL-S should be considered in patients with an occlusive retinal vasculopathy, a personal or family history of neurologic symptoms during middle age, and no evidence of a predisposing systemic medical disorder or autoimmune condition. Although there is no proven treatment for this disorder, unrecognized RVCL-S can lead to inappropriate and invasive management in addition to having significant implications for the patients’ relatives.
Footnotes
Ethical Approval
This case report was conducted in accordance with the Declaration of Helsinki. The collection and evaluation of all protected patient health information was performed in a Health Insurance Portability and Accountability Act (HIPPA)–compliant manner.
Statement of Informed Consent
Informed consent was obtained prior to performing the procedure, including permission for publication of all photographs and images included herein.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.