
Abstract
Keywords
Introduction
The structure of the telomere consists of repeating nucleotides found at the terminal ends of chromosomes and is essential for maintaining genomic integrity. Although gradual shortening of telomeres with repeated cell division occurs normally, a group of genetic diseases known as short telomere syndromes shares a premature aging phenotype that is driven by cell senescence and can be associated with premature organ failure. 1 Manifestations include early graying of the hair, bone marrow failure, liver disease, and pulmonary fibrosis. 1 In general, malignancies limited to myelodysplastic syndrome/acute myeloid leukemia and squamous cell cancers are diagnosed in 10% to 15% of adults. 2 Two short telomere disorders, Revesz syndrome and Coats plus syndrome, exhibit additional features of bilateral exudative retinopathy.3–5
To our knowledge, the onset of retinopathy associated with these diseases has been seen previously in children only. Here, we describe 2 patients with adult-onset presentations of retinopathy associated with short telomere syndromes.
Methods
This small retrospective case series included 2 adult patients who were seen at the Johns Hopkins University School of Medicine Department of Ophthalmology with retinopathy associated with short telomere syndromes. The workup included an ophthalmic examination, ocular imaging, telomere length measurement, and genetic testing. Both patients provided written informed consent for publication of their information and images.
Results
Case 1
A 53-year-old man was referred for newly recognized retinal vascular abnormalities. At that time, the visual acuity (VA) on an Early Treatment Diabetic Retinopathy Study (ETDRS) chart with spectacle correction was 20/16 OU. Ocular abnormalities were limited to the retina, including bilateral retinal telangiectasias temporal to the macula and around the vascular arcades (Figure 1, A and B). The left eye also had a preretinal hemorrhage (Figure 1B). Fluorescein angiography (FA) showed leakage from the telangiectasias (Figure 1, C and D). No leakage resulting from neovascularization (NV) was noted in either eye.
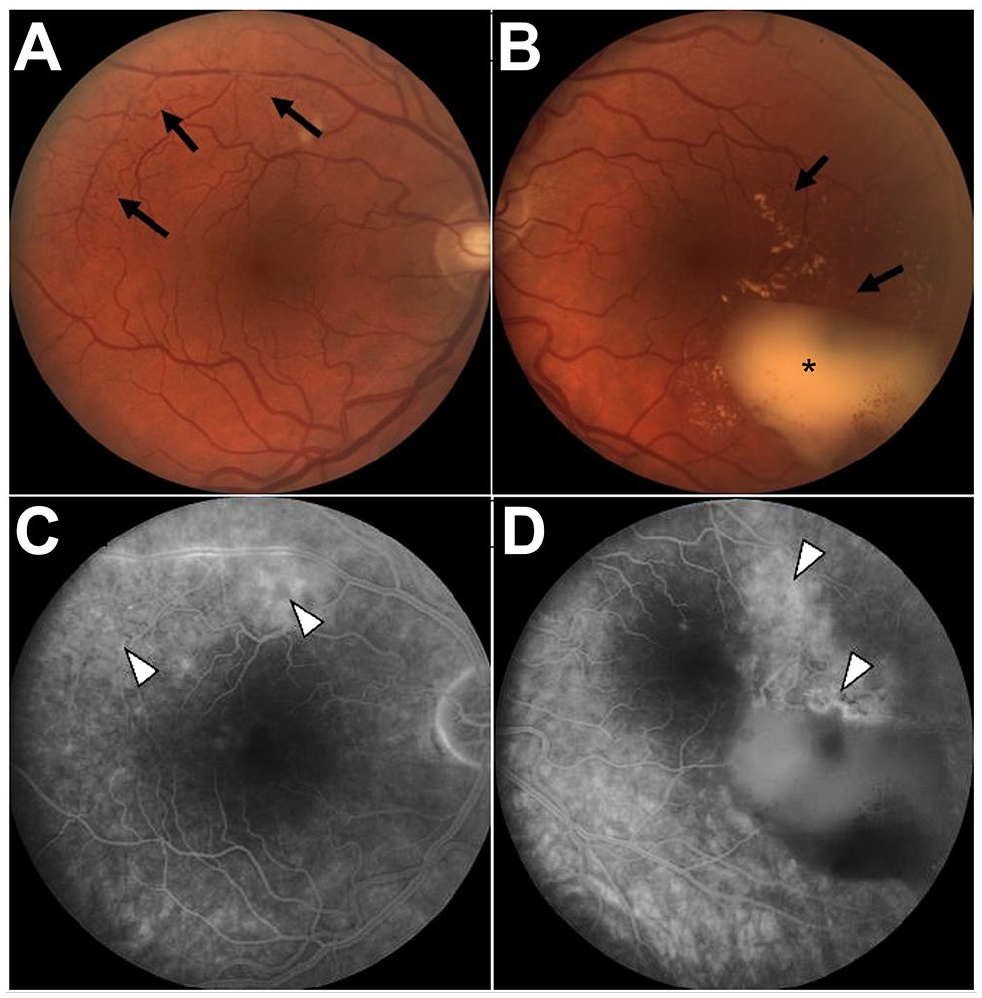
Fundus images from 2005 of the (A) right eye and (B) left eye. Retinal telangiectasia is noted in the left eye temporal/superotemporal to the macula and around the vascular arcades (black arrows) with preretinal hemorrhage (black asterisk). Late-phase fluorescein angiography of the (C) right eye and (D) left eye shows leakage from telangiectasias (white arrowheads).
Over 8 years of follow-up, preretinal and vitreous hemorrhages developed and spontaneously resolved twice in the left eye without retinal NV elsewhere. Subsequently, the patient developed a new vitreous hemorrhage in the left eye in the setting of a combined tractional rhegmatogenous retinal detachment without NV elsewhere. The patient had a successful repair of the detachment, during which fibrovascular proliferans was suspected, as noted in the operative note; however, it was not documented on photography.
Periodic retinal imaging over the next 7 years showed new telangiectatic vessels in both eyes. Repeat FA showed leakage from these telangiectatic vessels and areas of capillary nonperfusion but no NV elsewhere (Figure 2, A and B). In the absence of retinal NV, panretinal photocoagulation was not recommended. No cystoid thickening or abnormalities were seen on optical coherence tomography (OCT). Fifteen years after the patient’s presentation, an initial vitreous hemorrhage developed in the right eye.
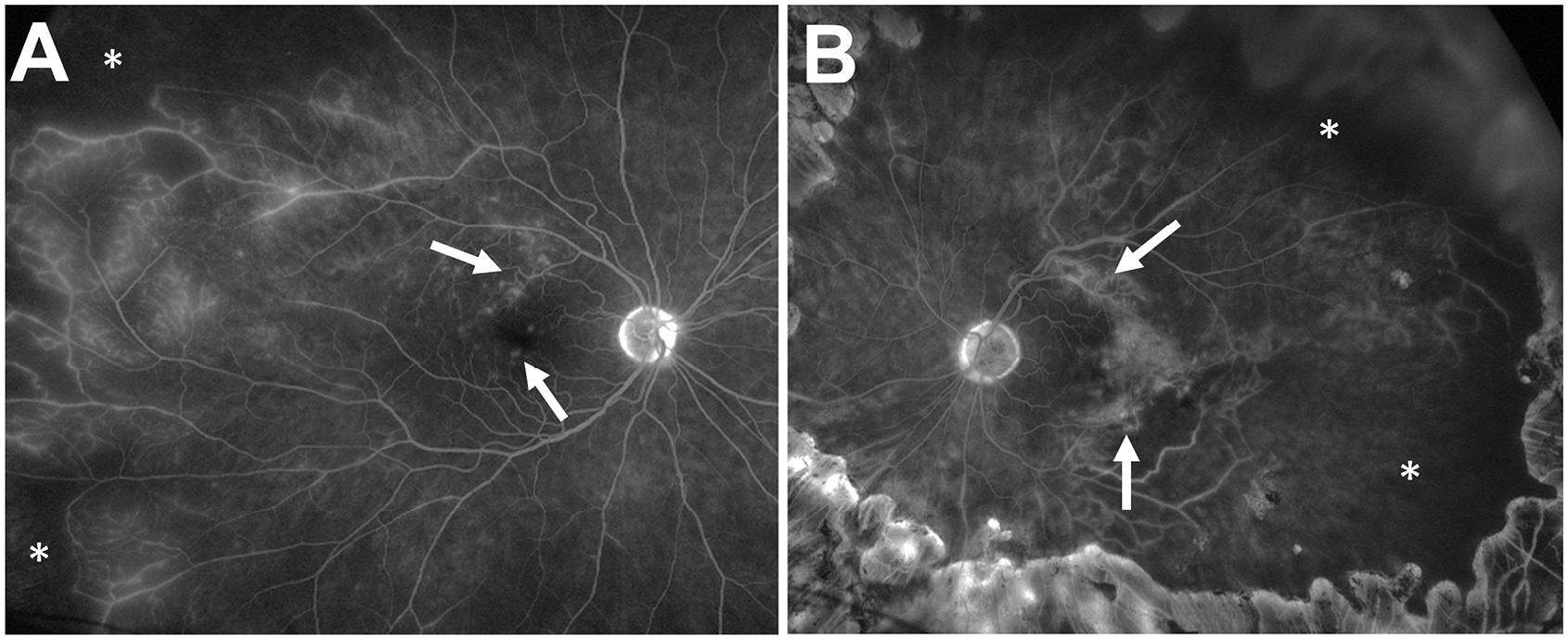
After 15 years of follow-up, late-phase ultra-widefield fluorescein angiography shows new telangiectatic vessels with leakage (white arrows) and areas of capillary nonperfusion (white asterisks) in the (A) right eye and (B) left eye.
Eight years after his initial presentation, the patient developed shortness of breath and abnormal skin lesions on the face. He was diagnosed with pulmonary fibrosis and facial basal cell carcinoma and was incidentally noted to have thrombocytopenia. Five years later, he was diagnosed with portal hypertension, hepatopulmonary syndrome with cirrhosis, and mild bone marrow failure and immunodeficiency.
Based on these manifestations, the patient was suspected to have a short telomere syndrome. His family history included a mother with no history of smoking who had pulmonary disease and 2 brothers with prematurely graying hair. The patient’s telomere length fell between the first and tenth percentile in lymphocytes (Figure 3), 6 and genetic testing confirmed a heterozygous missense variant in the RTEL1 gene: c.2114A>G, p.Tyr705Cys (NM_001283009).
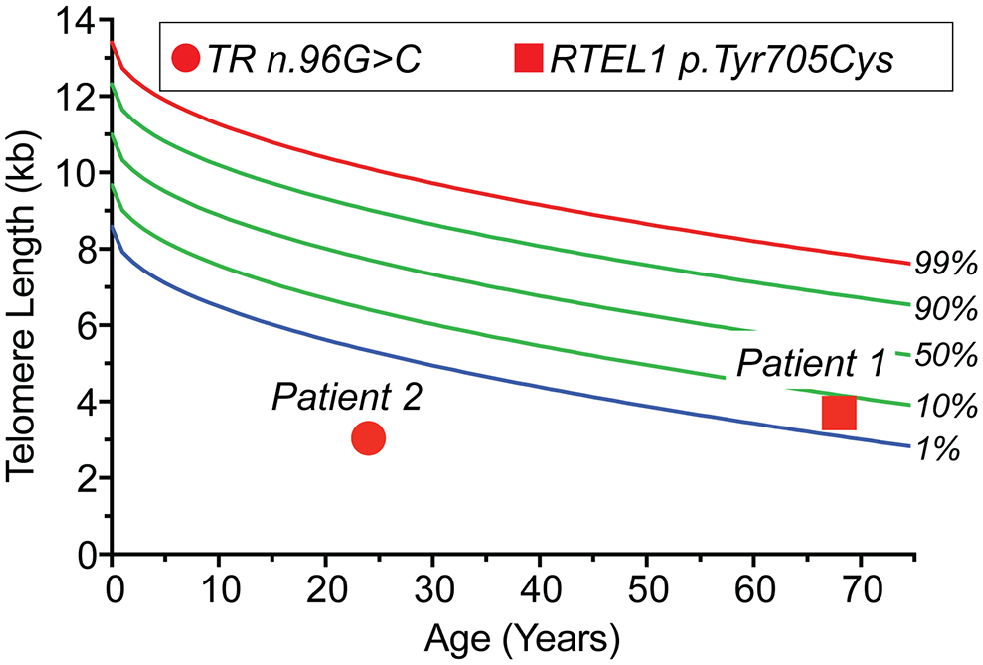
Telomere length and telomere gene mutations in 2 individuals with exudative retinopathy. The telogram shows lymphocyte telomere length by flow cytometry and fluorescence in situ hybridization for Case 1 and Case 2 relative to age. The data are plotted relative to a clinically validated nomogram 6 with percentile lines labeled on the right. Germline mutant gene and mutation are annotated in the key above.
The vitreous hemorrhage in the patient’s right eye was treated with 3 consecutive monthly intravitreal (IVT) aflibercept injections (extrapolated per protocols for proliferative retinopathy 7 ), resulting in an improvement in the VA to 20/20. The patient was followed at a multidisciplinary telomere center at Johns Hopkins, and the family was referred for genetic counseling. An evaluation found the patient to be ineligible for liver/lung transplantation, and he died at the age of 71 years from pulmonary fibrosis complicated by hepatopulmonary syndrome.
Case 2
A 26-year-old man with a history of floaters in the left eye since he was 20 years old was referred for evaluation of new floaters in the right eye. During this evaluation he was found to have abnormal retinal vasculature. Both he and his father had a known diagnosis of short telomere syndrome secondary to a single-nucleotide mutation in the gene encoding the telomerase RNA component, TERC (n.95G>C). The patient’s telomere length fell below the age-adjusted first percentile (Figure 3). He had no signs of short telomere syndrome until age 24 years, when he was found to have thrombocytopenia, macrocytosis, and a low CD4 count. He subsequently developed oral leukoplakia, progressive bone marrow failure, and T-cell immunodeficiency. Of note, his father had experienced multiple episodes of vitreous hemorrhage and was treated with peripheral scatter laser photocoagulation.
On examination, the patient’s VA on an ETDRS chart without correction was 20/20 OU. An anterior segment examination was normal, and a dilated fundus examination showed peripheral retinal capillary nonperfusion anterior to the equator in both eyes (Figure 4). There was a retinal telangiectasia with aneurysmal dilation of the capillaries surrounded by lipid and edema in the left eye. OCT showed normal macular architecture in both eyes. The patient was counseled on the risk for worsening retinopathy, and periodical follow-up was advised. The lipid and edema surrounding the area of the aneurysmal abnormality resolved with an additional 10 months, and subsequently 2 years, of follow-up (Figure 5), with no retinal NV or preretinal or vitreous hemorrhage. At the time of this writing, the patient was being evaluated for hematopoietic stem cell transplantation for the combined bone marrow failure–immunodeficiency phenotype.
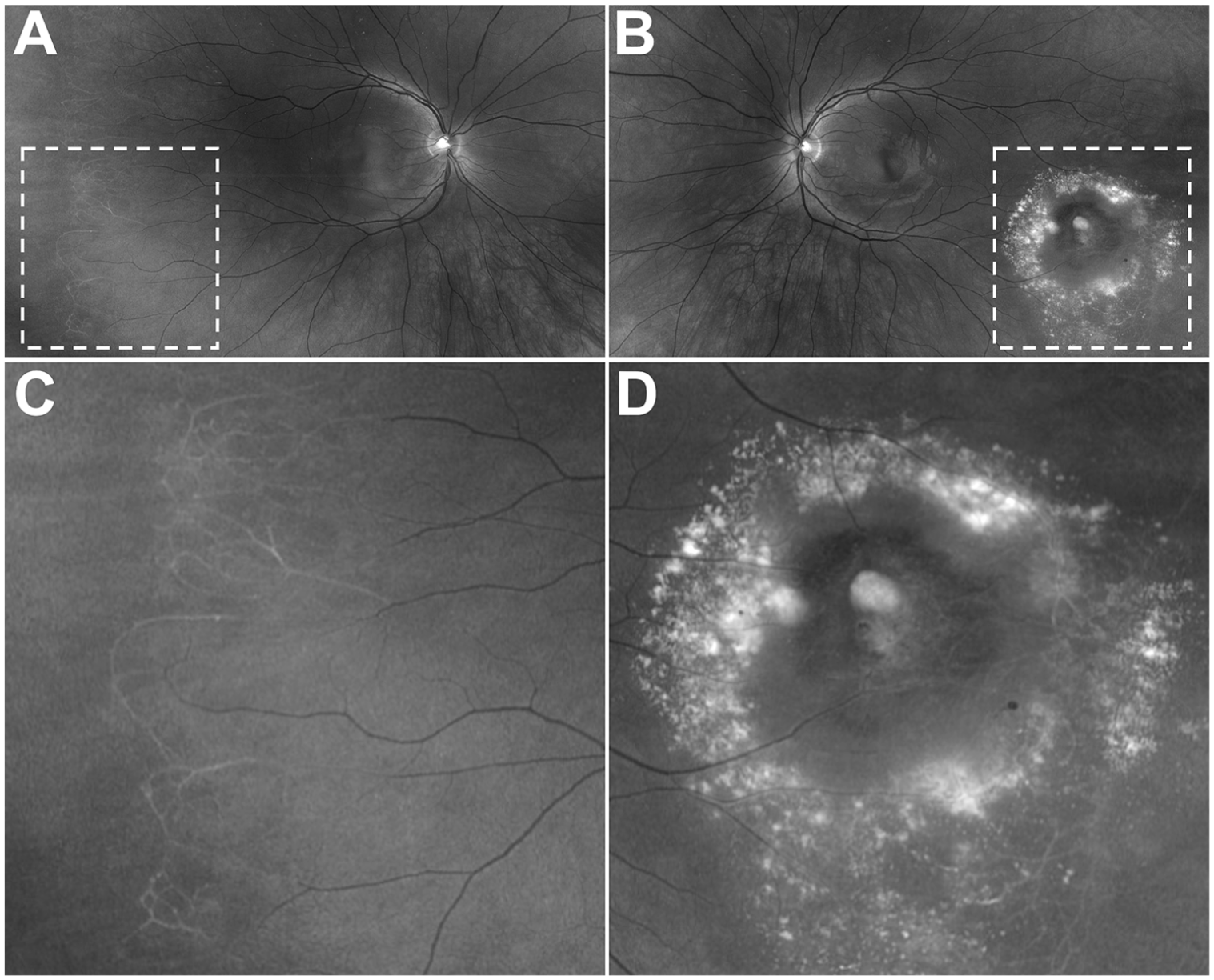
Fundus images (red-free) of the (A) right eye and (B) left eye. (C) A magnified image of peripheral nonperfusion with sclerotic-appearing vessels in the right eye. (D) A magnified image of a retinal telangiectasia, aneurysmal lesion, and surrounding lipid in the left eye. (Color figures available online.)
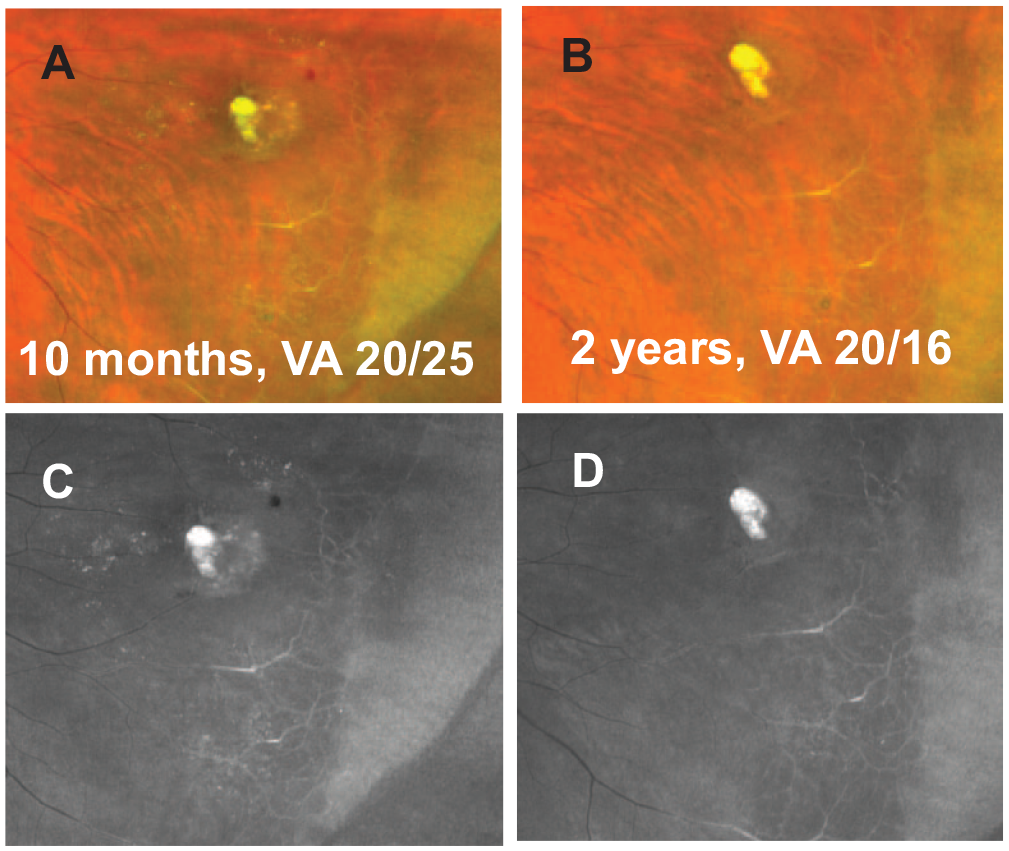
Fundus images of the left eye 10 months after initial presentation (A, color; B, red-free) and 2 years after initial presentation (C, color; D, red-free).
Conclusions
Retinopathy associated with short telomere syndromes can present in adulthood as incidental findings in the absence of vision loss, as shown in this case series. In Case 1, bilateral retinal telangiectasias with capillary nonperfusion and recurrent vitreous hemorrhage were heralding findings of a short telomere syndrome. In Case 2, the diagnosis was suspected because of family history; however, retinal changes were not reported until adulthood. Even then, the patient presented reporting nonspecific floaters without loss of VA concomitant with retinal telangiectasia and exudation temporal to the central macula in 1 eye and peripheral retinal capillary nonperfusion was documented on ultra-widefield imaging in both eyes. These cases extend the short telomere retinal manifestations seen in children with Revesz syndrome and Coats plus syndrome to adult-onset presentations that co-occur with pulmonary and liver disease as well as bone marrow failure and immunodeficiency (Table 1).2–5
Spectrum of Retinal Manifestations of Short Telomere Syndromes by Presentation: Children Compared With Adults.

The broad age spectrum at which retinopathy develops is consistent with the diverse clinical manifestations of short telomere syndromes. A milder retinal phenotype may be consistent in patients with mutations in TERT, RTEL1, and TERC, in which almost 90% of cases manifest in adulthood.1,6 Vascular abnormalities and perivascular iron deposition in the liver in patients with short telomere–mediated disease have been previously documented and have been hypothesized to be related to endothelial senescence. 8 In these cases, hepatopulmonary syndrome and portal vascular telangiectasia are also found; liver transplantation reverses this pathology.8,9 Vascular ectasia with gastrointestinal bleeding is also seen in pediatric disease, although some of these cases may be secondary to portal hypertension. 10 These observations support a vascular phenotype as a manifestation of the short telomere syndrome phenotype with systemic and retinal presentations. Long-term follow-up is important because progression of the retinopathy or complications (eg, vitreous hemorrhage) can be expected.2,3,6
To test for these conditions, one of many clinically available panels that include the telomere maintenance genes as well as others known to be associated with the disease can be ordered in consultation with a genetics provider. Clinical testing of the telomere length is also available, and the gold standard relies on flow cytometry–fluorescence in situ hybridization. 6 In patients with a high index of suspicion based on the personal history, family history, or both, the 2 studies may be ordered simultaneously. Although the telomere length measurement may help with variant interpretation, a pathogenic mutation may not be identifiable in approximately 20% of cases. 1
Treatment with panretinal photocoagulation or IVT antivascular endothelial growth factor injection may have been effective in Case 1, despite conclusive evidence of NV elsewhere. Case 2 showed that in the absence of neovascular complications or hemorrhage, peripheral nonperfusion in short telomere syndromes can be observed without treatment, similar to diabetic retinopathy. Last, the term telomere telangiectasia is proposed to denote characteristic retinal vascular abnormalities in the context of short telomere pathology, which can present as a spectrum of findings in children (Table 1), who may present with unilateral abnormalities and return for follow-up as an adult. 11
Although this case series cannot determine a cause-and-effect relationship between a short telomere molecular defect and retinal findings, at certain thresholds the short telomere molecular defect signals a DNA damage response that provokes cell senescence and apoptosis. One hypothesis is that this short telomere–mediated senescence signals endothelial cell senescence, provoking vascular ectasia. 8 This hypothesis is supported by some individuals with the short telomere syndrome phenotype who also develop vascular ectasia in the liver and, in such cases, perivascular iron deposition, suggesting evidence of a previous microscopic hemorrhage. 8 The retinal abnormalities noted in this case series support the investigation of hypotheses in this area, which may uncover additional insights into the disease’s pathophysiology.
Drawing conclusions about the prevalence of vascular abnormalities in adults with short telomere syndromes in this study is limited by the small sample and its retrospective nature. Future studies are needed that investigate the prevalence of retinopathy in this population and whether the retinal phenotype is an antecedent of disease and warrants genetic evaluation.
Footnotes
Ethical Approval
This case series was conducted in accordance with the Declaration of Helsinki. The collection and evaluation of all protected patient health information were performed in a US Health Insurance Portability and Accountability Act–compliant manner.
Statement of Informed Consent
Informed consent, including permission for publication of all photographs and images included herein, was obtained before the procedure was performed.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of the article: Dr. Nanegrungsunk received a research grant to Chiang Mai University from Roche. Dr. Bressler received grants to Johns Hopkins University from Bayer, Biogen, Genentech (Roche), Regeneron, and Samsung Bioepis and has a contract with the American Medical Association as editor-in-chief of JAMA Ophthalmology. None of the other authors declared potential conflicts of interest with respect to the research, authorship, and/or publication of the article.
Funding
Funding was provided by unrestricted philanthropic grants to the Johns Hopkins University School of Medicine and a grant from the National Institutes of Health (R01CA225027) to Dr. Armanios.