
Abstract
Introduction
The phenotypic heterogeneity of retinitis pigmentosa (RP) presents a significant challenge in establishing accurate diagnostic and therapeutic strategies. RP can present as an isolated ocular condition or in association with systemic manifestations, as observed in conditions such as Usher syndrome. 1 The documentation of additional clinical presentations remains valuable, as it contributes to a deeper understanding of the phenotypic spectrum of RP and helps elucidate the underlying pathophysiology.
Pathogenic variants in heparan-α-glucosaminide N-acetyltransferase (HGSNAT) result in autosomal recessive mucopolysaccharidosis type IIIC, also known as Sanfilippo syndrome.2,3 HGSNAT encodes a membrane-bound enzyme responsible for the transmembrane acetylation of heparan sulfate, a critical step in its lysosomal degradation. 4 Thus, pathogenic variants in HGSNAT result in the accumulation of unacetylated heparan sulfate within lysosomes.
Sanfilippo syndrome is a severe lysosomal storage disorder that often results in death by the third decade of life. 5 The condition is characterized by progressive degeneration of the central nervous system, often manifesting with developmental delay and behavioral disturbances, including hyperactivity. Within 5 to 10 years of symptom onset, patients typically experience a severe decline in cognitive and motor functions, often progressing to a vegetative state that persists until death.6,7
Two case series and several case reports have described nonsyndromic RP associated with pathogenic variants in the HGSNAT gene.8–10 Schiff et al 11 reported a genotype–phenotype correlation analysis in individuals with nonsyndromic retinopathy due to HGSNAT variants, expanding the phenotypic and genotypic spectrum of the condition through the identification of 3 new alleles. In their cohort, all but 2 patients presented with adult-onset, slowly progressive degeneration of the mid-peripheral retina. More recently, Pennesi et al 12 described the high phenotypical heterogeneity of HGSNAT-related retinopathy by describing late-onset retinopathy in patients harboring the c.1843G>A (p.Ala615Thr) pathogenic variant.
Building on these findings, our case series further contributes to the understanding of HGSNAT-related RP by detailing the genotypic and phenotypic features of 3 patients. Early recognition of this genetic subtype is crucial, as it not only facilitates accurate diagnosis and genetic counseling but also enables the timely identification of candidates for future therapeutic approaches and potential enrollment in clinical trials.
Methods
This study was approved by the Institutional Review Board of Johns Hopkins School of Medicine and conformed to the tenets of the Declaration of Helsinki. Due to the retrospective nature of the study, the requirement for obtaining informed consent was waived, and de-identified patient data were collected through chart review. Electronic medical records of eligible patients with a confirmed diagnosis of RP seen at the Genetic Eye Disease Center at the Wilmer Eye Institute up to January 2025 were screened for inclusion. Only patients harboring pathogenic HGSNAT variants were included. Exclusion criteria included age younger than 18 years and the presence of conditions other than the one under investigation. All patients underwent baseline clinical ophthalmologic examination, including multimodal imaging and genetic testing. Genetic analysis was performed using panel-based sequencing for inherited retinal diseases, and variants were classified according to the pathogenicity criteria established by the American College of Medical Genetics and Genomics. A comprehensive review of the system, including assessment for neurologic signs, was performed. None of the patients reported any neurologic signs or symptoms.
Case Series
Case 1
A 66-year-old White man was referred for evaluation of visual field defects. The patient reported progressive difficulty with night driving and poor peripheral vision in recent years. Family history was notable for poor peripheral vision loss in his mother, although no formal retinal diagnosis had been established (Figure 1, Case 1). Best-corrected visual acuity (BCVA) was 20/20 OD, intraocular pressure was within normal limits, and anterior segment examination was unremarkable. Fundus examination revealed optic nerve pallor, attenuated retinal vessels, peripapillary atrophy, and mild pericentral pigmentary changes in both eyes (Figure 2A). Fundus autofluorescence (FAF) demonstrated extensive areas of hypoautofluorescence in a peripapillary and pericentric distribution and an inner hyperautofluorescent ring surrounding the fovea (Figure 2B). Optical coherence tomography (OCT) demonstrated loss of the photoreceptor layer with preserved ellipsoid zone at the fovea (Figure 2C). Goldmann visual field testing demonstrated constricted isopters to all stimuli, with a pericentral area of visual field loss in both eyes (Figure 2D). Full-field electroretinography (ERG) revealed moderate generalized dysfunction of rod and cone photoreceptors in both eyes (Figure 2, E and F). Genetic testing revealed 2 disease-causing variants in the HGSNAT gene: c.1843G>A (p.Ala615Thr) and c.1102A>T (p.Lys368*).
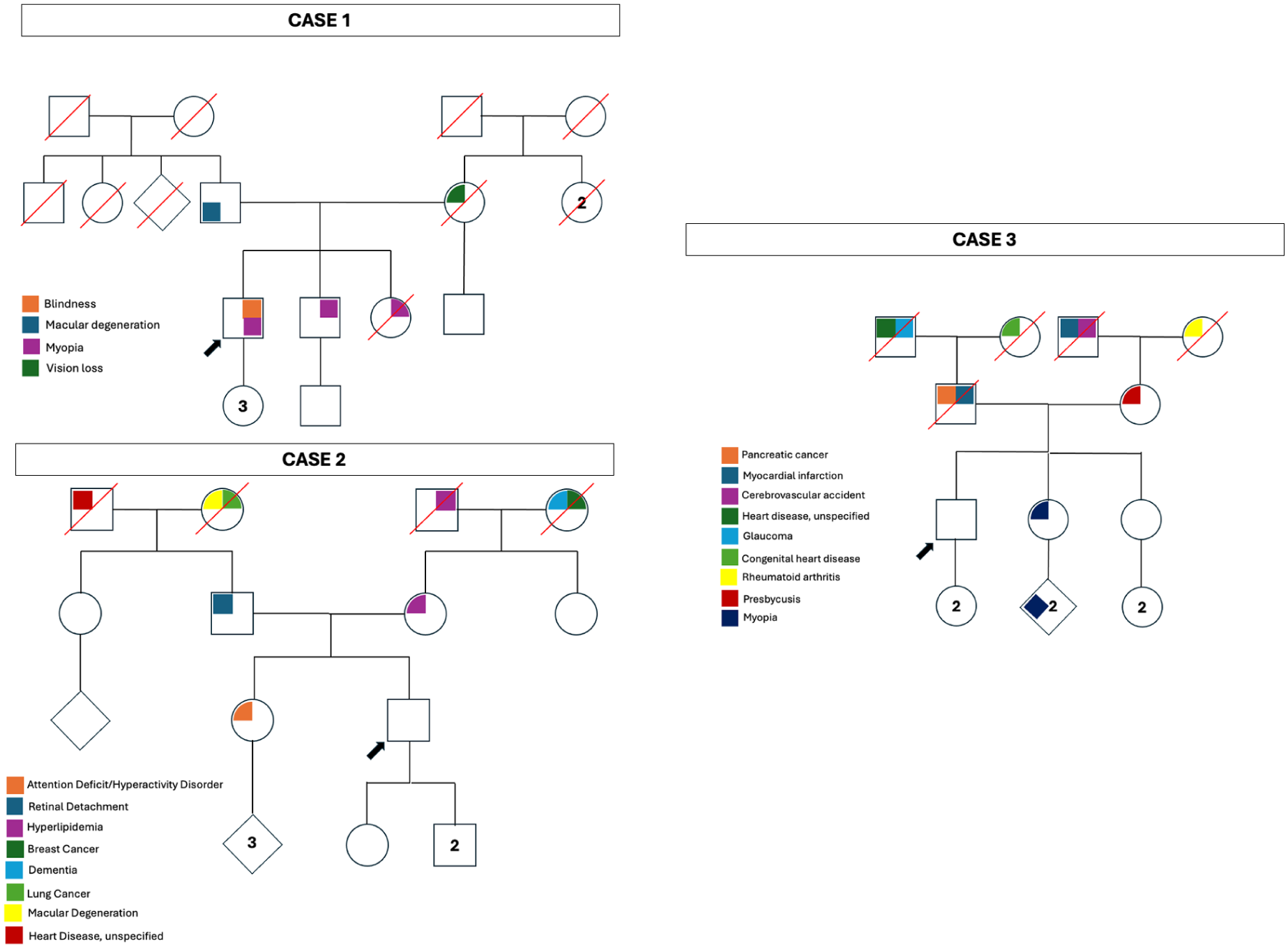
Pedigrees of the patients in Cases 1–3. None of the pedigrees demonstrated a family history of retinitis pigmentosa or childhood neurodegenerative diseases. In all cases, segregation analysis was performed to confirm variant pathogenicity. Numbers within pedigree symbols indicate multiple offspring.
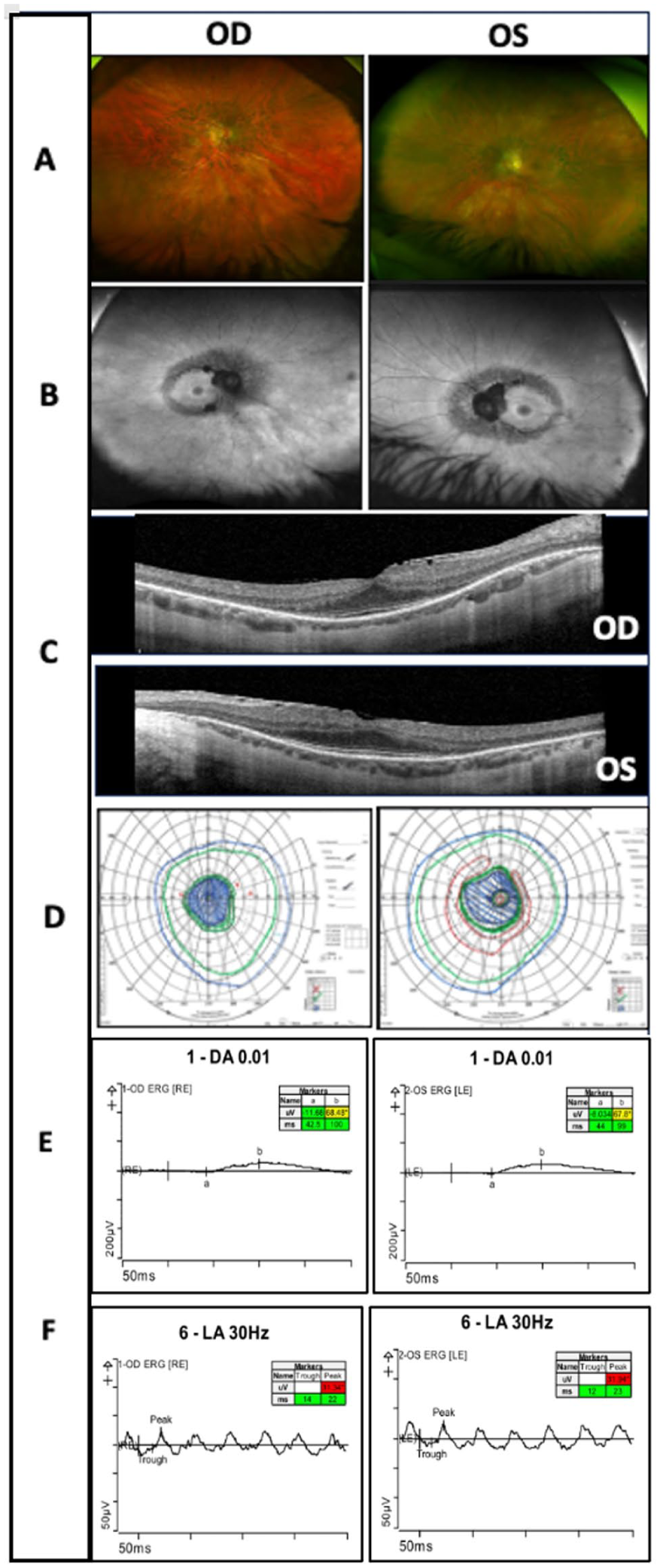
Multimodal imaging findings for Case 1. (A) Color fundus photographs demonstrating attenuated retinal vessels, optic nerve pallor, and pigmentary changes in a pericentric distribution in both eyes. (B) Fundus autofluorescence images revealing areas of hypoautofluorescence in a pericentral distribution, consistent with photoreceptor and retinal pigment epithelium dysfunction. (C) Optical coherence tomography demonstrating photoreceptor loss with preserved ellipsoid zone at the fovea in both eyes. (D) Goldmann visual field testing showing constricted isopters to all stimuli with pericentral areas of visual loss in both eyes. Full-field electroretinography demonstrating (E) reduced 0.01 dark-adapted responses and (F) abnormal 30 Hz light-adapted flicker responses, consistent with moderate generalized dysfunction of the rod and cone photoreceptor system in both eyes.
Case 2
A 39-year-old White man with no significant past medical or ocular history presented with a 3-month history of subtle flashes and scotomas that were more pronounced at night. There was no history of consanguinity in his pedigree (Figure 1, Case 2). Symptoms initially involved the left eye and later progressed to involve both eyes. The patient also reported intermittent spasmodic pain in the suprapubic region and 1 episode of leg weakness with a similar temporal onset. Systemic evaluation for autoimmune retinopathy and uveitis was negative, except for elevated cerebrospinal fluid oligoclonal bands.
At presentation, BCVA was 20/20 OU. Intraocular pressure, anterior segment examination, and dilated fundus examination were unremarkable (Figure 3A). FAF demonstrated concentric hyperautofluorescent rings in both eyes, raising suspicion for RP (Figure 3B). OCT demonstrated preservation of the ellipsoid zone in the foveal region with parafoveal retinal thinning (Figure 3C). Previously performed full-field ERG confirmed severe photoreceptor dysfunction; however, the tracings were unavailable for review.
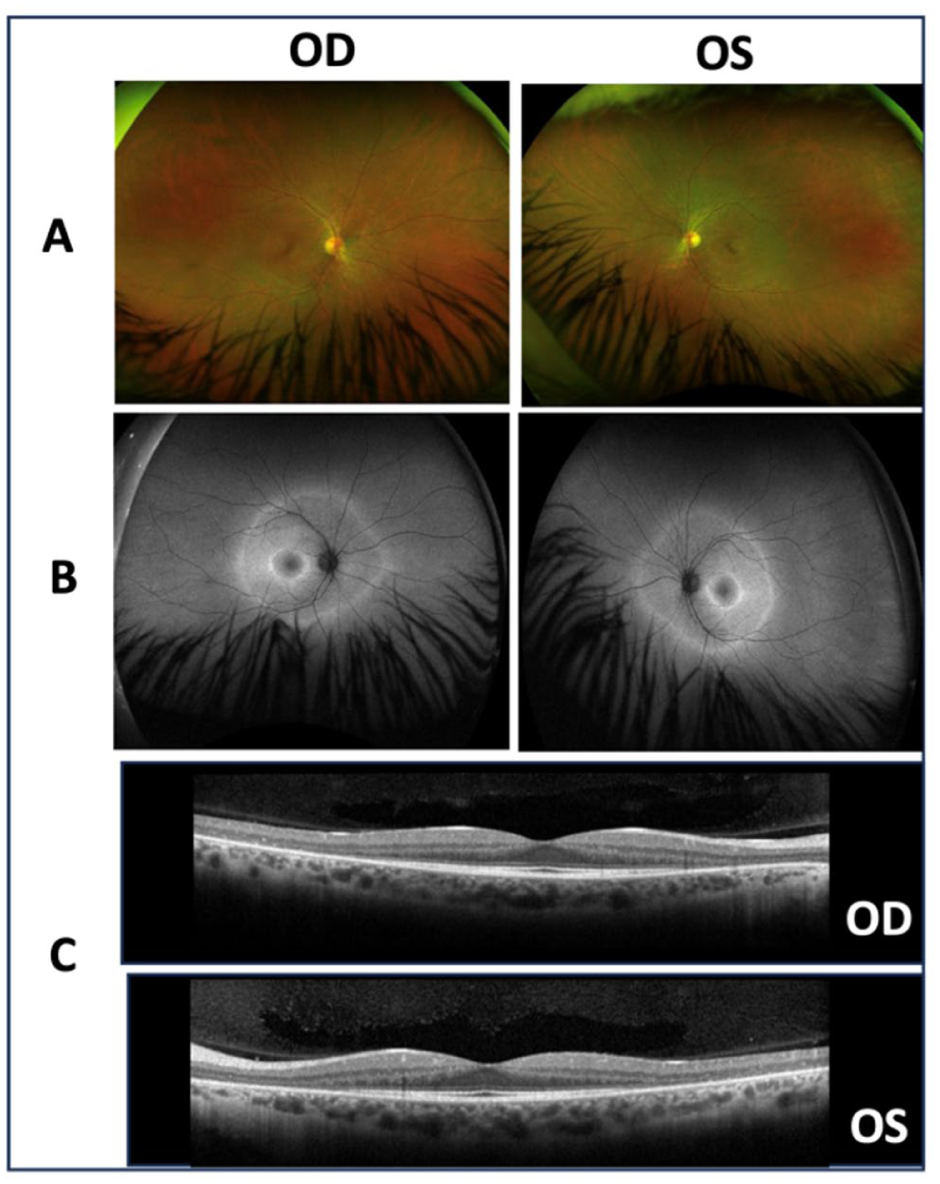
Multimodal imaging findings for Case 2. (A) Color fundus photographs demonstrating no significant abnormalities. (B) Fundus autofluorescence images revealing concentric hyperautofluorescent rings in the macula and beyond the vascular arcades in both eyes. (C) Optical coherence tomography demonstrating preservation of the ellipsoid zone line in the foveal region with parafoveal retinal thinning.
Given the imaging findings highly suggestive of RP, genetic testing was performed, revealing homozygous HGSNAT c.1843G>A (p.Ala615Thr) pathogenic variants.
Case 3
A 51-year-old White man presented for evaluation 5 years after being diagnosed with occult macular dystrophy after a multifocal ERG demonstrated impaired cone photoreceptor function in both eyes. At presentation, his BCVA was 20/100 OD and 20/125 OS. Intraocular pressure, anterior segment examination, and dilated fundus examination were unremarkable (Figure 4A). FAF demonstrated peripheral hypoautofluorescent patches and a faint parafoveal hyperautofluorescent ring (Figure 4B). OCT revealed diffuse parafoveal atrophy (Figure 4C). Full-field ERG findings were consistent with severe generalized cone and rod photoreceptor dysfunction in both eyes, with decreased B-wave amplitudes and reduced flicker responses compared with normal values (Figure 4D). Genetic testing revealed a heterozygous pathogenic variant in the HGSNAT gene: c.1843G>A (p.Ala615Thr).
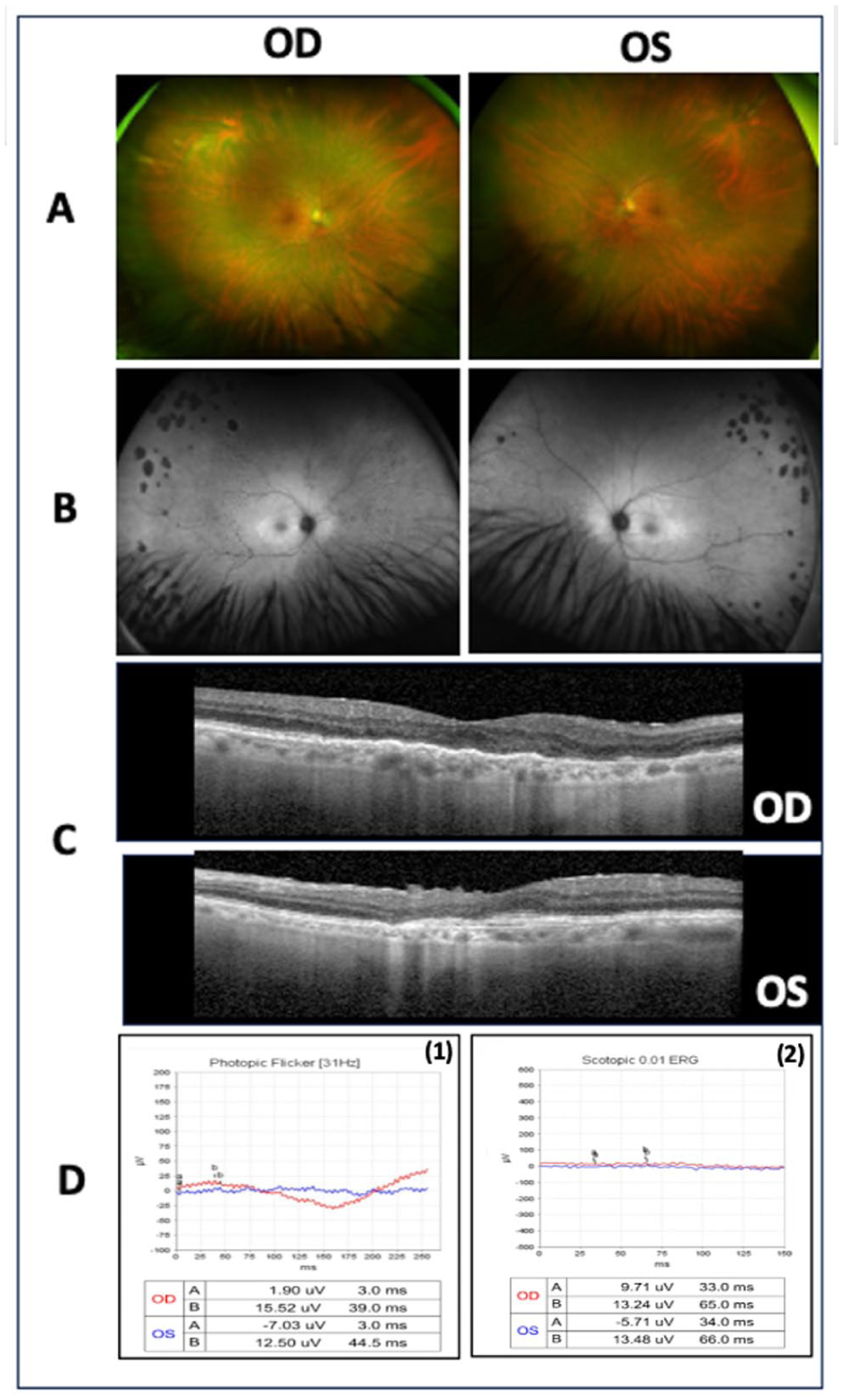
Multimodal imaging findings for Case 3. (A) Color fundus photographs demonstrating no significant abnormalities. (B) Fundus autofluorescence images revealing patchy peripheral areas of hypoautofluorescence and a faint parafoveal hyperautofluorescent ring in both eyes. (C) Optical coherence tomography demonstrating diffuse parafoveal retinal atrophy in both eyes. Full-field electroretinography demonstrating (D1) markedly reduced 30 Hz light-adapted flicker responses and (D2) severely diminished 0.01 dark-adapted responses, consistent with severe generalized dysfunction of the rod and cone photoreceptor system in both eyes, with reduced B-wave and flicker amplitudes compared with normal values.
Conclusions
Our case series highlights the broad ocular phenotypic spectrum associated with the HGSNAT c.1843G>A (p.Ala615Thr) and c.1102A>T (p.Lys368*) pathogenic variants. Interestingly, all 3 patients presented with at least 1 Ala615Thr pathogenic variant but demonstrated markedly variable ocular manifestations, demonstrating the wide clinical heterogeneity associated with this genotype.
The first patient exhibited classic signs of RP supported by functional testing, despite a BCVA of 20/20 at presentation. Posterior pole and peripheral retinal findings and visual field defects were compatible with RP. Genetic testing identified 2 pathogenic HGSNAT variants. The c.1843G>A (p.Ala615Thr) variant, a mild hypomorphic allele, is the most common variant described in case series of nonsyndromic HGSNAT-related RP and is typically associated with late-onset, slowly progressive disease.11,12 The c.1102A>T (p.Lys368*) variant, which results in a premature stop codon, has not previously been described and may account for the more typical RP phenotype observed in this patient.
The second patient harbored biallelic HGSNAT c.1843G>A (p.Ala615Thr) pathogenic variants but demonstrated a markedly different clinical presentation, with no visible retinal lesions on examination and only mild ocular symptoms. In this case, FAF and full-field ERG were crucial to identify retinal dysfunction. Notably, the patient underwent extensive evaluation to rule out autoimmune retinopathy, acute zonal occult outer retinopathy, and other infectious or inflammatory etiologies before referral to our inherited retinal disease clinic and subsequent genetic testing.
The third patient presented with the most severe phenotype, with reduced BCVA at presentation, peripheral hypoautofluorescent lesions despite a normal fundus examination, and extensive parafoveal atrophy on OCT. Genetic testing identified a heterozygous c.1843G>A (p.Ala615Thr) pathogenic variant, suggesting the possibility of additional undetected variants or modifying factors influencing disease severity.
The phenotypic diversity observed in these 3 cases underscores the importance of investigating HGSNAT-associated retinopathy and identifying genetic modifiers that influence clinical presentation.
Previously, Schiff et al 11 had reported the clinical and functional findings of 16 individuals with biallelic HGSNAT variants and isolated RP. Most patients presented with adult-onset progressive degeneration of the mid-peripheral retina associated with nyctalopia and visual field constriction. Interestingly, the authors identified 7 novel sequence variants and 4 previously reported mucopolysaccharidosis type IIIC-associated variants occurring in trans with the hypomorphic p.Ala615Thr allele, thereby expanding the genotypic spectrum of HGSNAT-associated retinopathy. Moreover, the presence of a homozygous (p.Ala615Thr) variant in both an affected patient and their unaffected 73-year-old sibling suggested a possible role for trans-acting genetic or environmental modifiers that influence the retinal phenotype. In that analysis, the retinal phenotype was most commonly associated with a pericentral symmetric distribution, similar to the findings observed in our first case. 11
A detailed phenotypic description of HGSNAT-associated retinopathy was reported by da Palma et al, 12 who noted high phenotypic heterogeneity in their series. Interestingly, in their cohort, all 11 patients with the c.1843 G>A (p.Ala615Thr) variant presented with late-onset retinopathy, in contrast to those not carrying this variant. 12 The c.1843 G>A (p.Ala615Thr) variant was the most frequently identified allele due to the high prevalence of this single-nucleotide polymorphism in the general population. It was found to be homozygous in 3 patients and heterozygous in 5 patients.
Similarly, in the case series reported by Schiff et al, 13 patients presented with the c.1843 G>A (p.Ala615Thr) variant, including 8 heterozygous and 5 homozygous individuals. Cystoid macular edema (CME) was also observed in several cases, occurring in 4 patients carrying the c.1843 G>A (p.Ala615Thr) variant in the Schiff et al 11 cohort and in 4 patients overall in the da Palma et al 12 case series.
Previously, Long et al 9 had reported 2 novel HGSNAT variants associated with nonsyndromic RP in a Chinese family. The affected patients presented with bilateral peripheral retinal atrophy, widespread intraretinal bone spicule-like pigmentation, and CME. Both patients harbored 2 novel HGSNAT variants: c.1048C>T (p.Gln350*) and c.1908A>G (p.*636Trpext*12). Carrera et al recently described 2 cases of nonsyndromic RP presumably caused by compound heterozygous pathogenic variants in HGSNAT: c.1843G>A (p.Ala615Thr) and c.1565C>T (p.Thr522Met). These patients exhibited bilateral CME as well as retinal neovascularization. The authors classified c.1565C>T (p.Thr522Met) as a severe pathogenic variant, whereas c.1843G>A (p.Ala615Thr) was considered a hypomorphic allele. 8
More recently, Pennesi et al 12 reported a wide range of ophthalmologic findings, ranging from mild vascular attenuation to marked RPE atrophy and bone spicule pigmentation. Similarly, all 5 cases reported by Haer-Wigman et al 10 presented with nonsyndromic RP, with differing clinical symptoms and age of onset. Additional case reports have documented similar ophthalmic findings despite differing HGSNAT variants.8,9,13 Although several pathogenic variants have been implicated, the c.1843G>A (p.Ala615Thr) variant remains the most commonly reported allele in White patients with isolated HGSNAT-related RP.14,15 Our case series further supports this observation. Further studies are warranted to understand the role of trans-acting genetic and environmental modifiers that may influence retinal phenotype expression.
Our study has several limitations, including its retrospective nature and the small number of patients meeting eligibility criteria. Nevertheless, given the limited number of reported cases with HGSNAT pathogenic variants, this case series provides valuable insights into the expanding genotypic and phenotypic spectrum of isolated HGSNAT-related RP. Notably, 2 patients in this case exhibited normal fundus findings on clinical examination. However, FAF revealed patterns of retinal dysfunction consistent with RP. These findings underscore the importance of considering HGSNAT-related RP in the differential diagnosis of patients with normal or subtle retinal findings and symptoms. Moreover, early recognition is essential in the context of clinical trials, as timely and accurate diagnosis may facilitate access to potential therapeutic interventions.
Footnotes
Ethical Considerations
This study was approved by the Institutional Review Board of Johns Hopkins School of Medicine and conformed to the tenets of the Declaration of Helsinki.
Consent to Participate
Due to the retrospective nature of the study, the requirement for informed consent was waived, and de-identified patient information was collected through chart review.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr. Ahmed received funding from the Retina Rising Professorship.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.