
Abstract
Cabergoline, a dopamine receptor agonist, has shown beneficial effects as a preventive treatment for migraine, with no serious adverse events reported. This study aims to evaluate whether cabergoline is superior to placebo in reducing monthly migraine days (MMD), while assessing safety and tolerability. In a randomized, parallel-group, placebo-controlled, double-blind superiority phase II trial, 150 adults with 4–14 MMD will be included. Participants are randomized (1:1:1) to cabergoline 0.5 mg, cabergoline 1.0 mg or placebo once weekly as add-on treatment for 12 weeks. In a 12-week open-label extension, all participants, including those initially assigned to placebo, will be re-randomized (1:1) to cabergoline 0.5 mg or 1.0 mg once weekly to assess sustained effects and safety. The primary outcome is change in MMD from baseline to the final four weeks of the double-blind phase. Key secondary outcomes include ≥50% responder rate, change in the number of moderate-to-severe headache days, acute medication use and patient-reported outcomes (PGIC, HIT-6, MIDAS, WPAI). Exploratory analyses include biomarkers, pharmacogenetics and cost-effectiveness. Analyses will follow the intention-to-treat principle.
This is a visual representation of the abstract.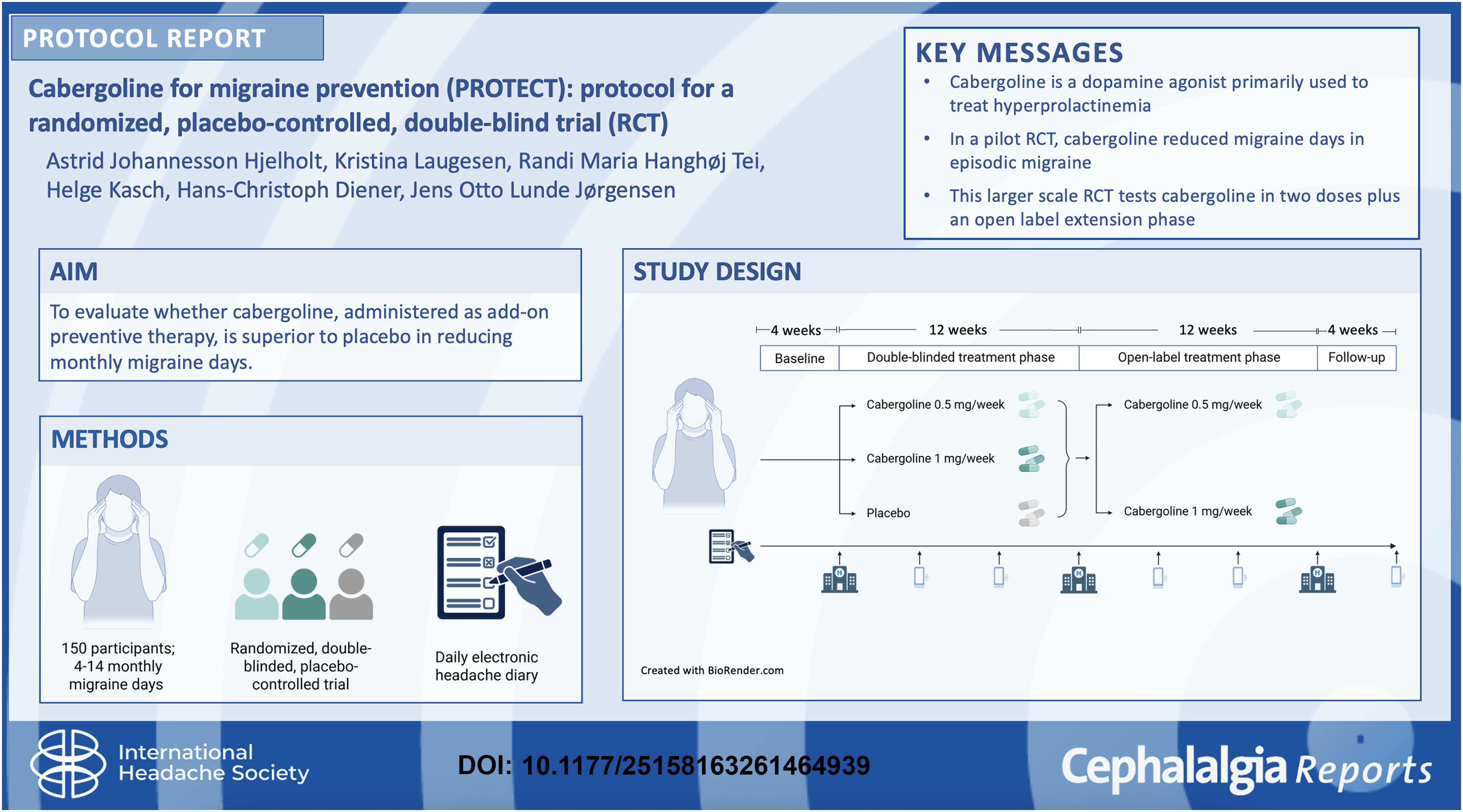
Introduction
Migraine is a complex neurovascular disorder with an estimated global lifetime prevalence of 17.5%, which makes it the second leading cause of disability worldwide, quantified as years lived with disability.1–3 Although several preventive treatments exist, including newer calcitonin gene-related peptide (CGRP) antagonistic compounds, many patients remain undertreated due to either affordability, suboptimal efficacy or tolerability issues.3–5 Therefore, there is a continued need for preventive treatments that are both effective and affordable.
Activation of the trigeminovascular system and release of neuropeptides such as CGRP are central to migraine pathophysiology.3,6 Emerging evidence also implicates prolactin. 7 Elevated serum prolactin levels have been reported in individuals with migraine,8,9 and migraine-like headache is frequent in hyperprolactinaemia. 9 Prolactin receptors are expressed on trigeminal sensory neurons, 10 and experimental data suggest that prolactin increases neuronal excitability, providing a potential mechanistic link 11 (Figure 1).
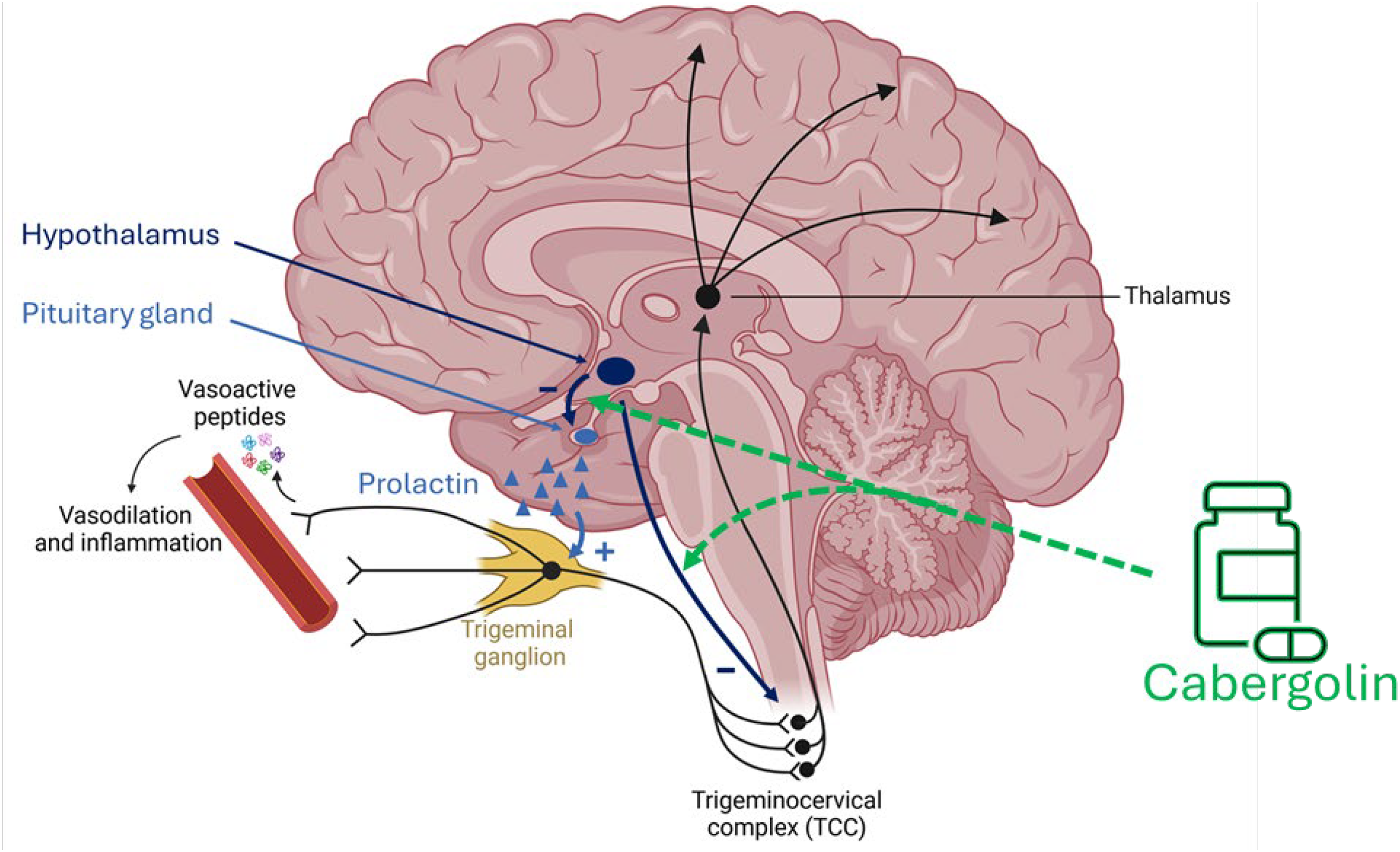
Proposed mechanisms of cabergoline in migraine. Activation of the trigeminovascular system is central to migraine pathophysiology. Prolactin receptors are expressed on trigeminal sensory neurons, and prolactin may enhance neuronal excitability. Dopaminergic signalling inhibits prolactin secretion and modulates nociceptive processing. Cabergoline, a dopamine D2 receptor agonist, may reduce migraine activity through prolactin suppression and dopaminergic modulation within trigeminovascular pathways.
Prolactin secretion is inhibited by dopamine, a neurotransmitter also implicated in central pain modulation.12,13 Dopaminergic signalling may explain several premonitory migraine symptoms, such as yawning and somnolence,14,15 while experimental models suggest an antinociceptive effect of dopamine within the trigeminovascular system.12,14,15
Cabergoline is a dopamine D₂ receptor agonist approved for the treatment of hyperprolactinaemia.16,17 It also displays affinity for serotonin 5-HT₁B/₁D receptors and has pharmacokinetic properties that permit once-weekly dosing with generally good tolerability.16,17
Dopamine agonists, including cabergoline, improve prolactin-associated headache even in the absence of pituitary tumour shrinkage,18–22 and benefit has also been suggested in migraine without hyperprolactinaemia.23–25 In a recent randomized, placebo-controlled pilot trial, once-weekly cabergoline (0.5 mg) reduced monthly migraine days (MMD) and improved Patient's Global Impression of Change (PGIC), without serious adverse events, among patients with up to 14 MMD. 26 The effect size was comparable to established preventive therapies, including anti-CGRP agents. 27
The primary objective of this trial is to evaluate whether cabergoline, administered as add-on therapy, is superior to placebo in reducing MMD in patients with 4–14 MMD, excluding those with chronic daily headache or suspected medication-overuse headache. Safety and tolerability will be assessed as secondary outcomes. Placebo is used as a comparator to allow an unbiased assessment of efficacy and to ensure consistency with established standards in preventive migraine trials. 28
Methods
This study protocol is reported in accordance with the SPIRIT 2013 statement.
Trial design
The PROTECT trial is a prospective, randomized, double-blind, placebo-controlled, parallel-group trial with three distinct intervention groups: (1) cabergoline 0.5 mg/week, (2) cabergoline 1.0 mg/week and (3) placebo. After screening and informed consent, participants enter a 4-week baseline phase to document migraine frequency and confirm eligibility (4–14 MMD). Participants who continue to meet the inclusion criteria will be enrolled in a 12-week double-blind treatment phase. This is followed by a 12-week open-label treatment phase, in which all participants receive active treatment (cabergoline 0.5 or 1.0 mg). During the open-label phase, dose reduction from 1.0 to 0.5 mg weekly is permitted if tolerability issues occur. A final 4-week safety follow-up phase is included to monitor for any delayed adverse events, in accordance with International Headache Society guidelines. 28 A schematic overview is shown in Figure 2.
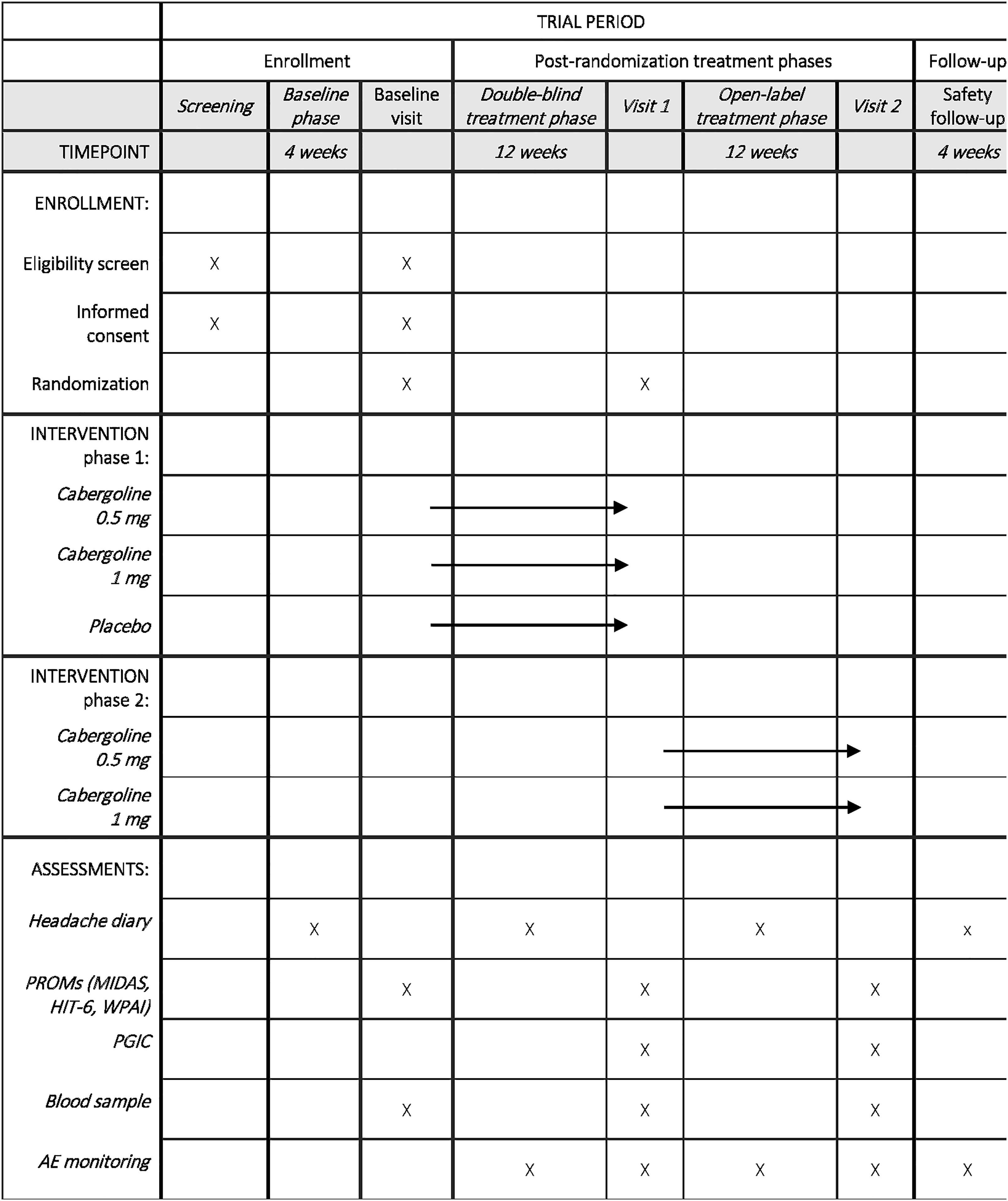
Participant timeline: schedule of enrolment, interventions and assessments. Overview of screening, randomisation, treatment phases and assessments conducted during the study, including headache diaries, patient-reported outcomes, blood sampling and adverse event monitoring.
Trial setting
The trial will be conducted at Aarhus University Hospital. All study visits, procedures and data collection will be carried out at the Department of Internal Medicine and Endocrinology, Aarhus University Hospital.
Study population
A total of 150 patients with 4–14 MMD will be enrolled. Eligibility criteria are listed in Table 1. Participants will be recruited via social media, patient organizations, general practices and neurology clinics. Recruitment will continue until the target sample size is reached, with progress monitored by the investigative team and strategies adapted if needed. No remuneration will be offered.
Eligibility criteria.

Intervention
During the double-blind phase, participants receive once-weekly cabergoline 0.5 mg, cabergoline 1.0 mg or matching placebo. In the subsequent open-label phase, all participants receive active cabergoline treatment (0.5 or 1.0 mg once weekly). Study medication is taken orally as add-on therapy to usual care. Participants may be treatment-naïve, previously treated or receiving preventive migraine medication. Preventive migraine treatments are permitted only if the regimen has remained stable for at least 3 months prior to baseline and throughout the trial period. Acute migraine medications are permitted as needed and recorded prospectively in the daily headache diary.
Participants self-administer the medication at home on the same weekday each week, preferably at bedtime. Adherence is supported through standardized instructions, digital reminders and capsule count at each visit.
Discontinuation may occur in case of intolerable adverse events, newly identified exclusion criteria, non-compliance or participant request. Discontinuations are documented with follow-up encouraged.
Outcomes
The primary outcome is the difference in mean change in MMD from baseline to the last 4 weeks of the 12-week double-blind treatment phase, based on daily electronic diary entries and defined as the number of days with migraine per 28-day period. This aligns with international guidelines for preventive migraine trials. 28 Reductions of 1–3 MMD have been reported for established preventives, and >1 MMD is considered clinically meaningful. 27
Secondary outcomes include the proportion of participants achieving ≥50% reduction in MMD, change in the number of moderate to severe headache days, use of acute migraine-specific medication, and patient-reported outcomes assessed by the Migraine Disability Assessment (MIDAS), 29 Headache Impact Test (HIT-6), 30 Work Productivity and Activity Impairment Questionnaire: Migraine (WPAI: Migraine) 31 and PGIC. 32 MIDAS, HIT-6 and WPAI: Migraine are validated instruments in migraine populations, and PGIC is a widely used single-item global assessment of treatment response. Secondary outcomes are assessed from baseline to the end of the double-blind treatment phase. The same outcomes will be collected at the end of the open-label phase and analysed descriptively as exploratory outcomes.
Open-label data will be analysed descriptively to explore dose-related trends without formal statistical comparisons. Safety and tolerability will be assessed throughout the trial, and persistence of effects during the open-label extension phase.
Exploratory outcomes include changes in serum prolactin, metabolic and inflammatory biomarkers, sex hormones, pharmacogenetic analyses of prolactin receptor and dopaminergic pathway variants and cost-effectiveness.
Study procedures
Participants complete a daily electronic headache diary during the baseline, double-blind and open-label treatment phases (Figure 2). The diary records headache characteristics, severity, associated symptoms, acute medication use and healthcare contacts, and serves as the primary source for MMD and other headache-related outcomes. Headache severity is recorded using a three-level categorical scale (mild, moderate, severe) based on functional impairment. Entries are submitted via a secure REDCap-based platform, with real-time recording to promote data completeness and minimize recall bias.
Participants attend in-person visits at week 0, 12 and 24. Venous blood samples are collected to assess serum prolactin, metabolic and inflammatory biomarkers (lipids, HbA1c, hs-CRP), and sex hormones. At baseline, additional samples are obtained for pharmacogenetic analyses of variants in prolactin receptor and dopaminergic pathway genes (DRD2/NCOR1, DBH and SLC6A3) implicated in migraine and cabergoline response.33,34
At each visit, participants complete MIDAS, HIT-6 and WPAI questionnaires; PGIC is assessed at weeks 12 and 24. Patient-reported outcomes are analysed as secondary outcomes at week 12 and descriptively at week 24.
Retention is supported through reminders, and outcome data will be collected where possible even if study treatment is discontinued.
Harms (adverse events)
Harms will be defined as adverse events and serious adverse events in accordance with Good Clinical Practice. Adverse events will be assessed at each study visit and scheduled telephone contact, with participants actively asked about any events since the previous contact. Participants will also be systematically assessed using a predefined symptom checklist covering respiratory, renal/retroperitoneal and cardiac symptoms. If relevant symptoms are reported, appropriate clinical evaluation and relevant diagnostic investigations will be performed as clinically indicated. Participants will be instructed to contact the investigator at any time in case of new or worsening symptoms. All adverse events will be recorded from informed consent until four weeks after completion of study treatment.
Symptoms suggestive of impulse control disorders will be systematically assessed using the Questionnaire for Impulsive–Compulsive Disorders in Parkinson's Disease – Rating Scale (QUIP-RS) at baseline, week 12, week 24 and week 28 to monitor the emergence or worsening of impulse control disorders. Clinically relevant findings will prompt further clinical evaluation and discontinuation of study treatment if indicated.
For women of childbearing potential, urine pregnancy testing will be performed at baseline and every four weeks. In case of a positive test, study treatment will be discontinued and the participant withdrawn and followed according to protocol-defined safety procedures.
Given the low dose and limited treatment duration (≤24 weeks), routine echocardiographic screening is not performed. Participants with a history of pulmonary, retroperitoneal or pericardial disorders, including heart valve disease, are excluded at baseline and cardiac auscultation is performed at in-person visits. Echocardiography will be undertaken if clinically indicated by symptoms or abnormal cardiovascular findings.
Randomisation and blinding
During the double-blind phase, randomisation will be performed by Glostrup Pharmacy using a computer-generated restricted sequence with varying block sizes, allocating participants in a 1:1:1 ratio to cabergoline 0.5 mg, cabergoline 1.0 mg or placebo. The allocation sequence will be held by the pharmacy, which will encapsulate, label and dispense identically appearing study medication. Participants, investigators, study staff and outcome assessors will be blinded throughout the double-blind phase.
Emergency unblinding is permissible in medical emergencies using sealed code-break envelopes. Final unblinding will occur only after all participants have completed the double-blind phase.
After completion of the double-blind phase, all participants, including those initially allocated to placebo, will be re-randomised in a 1:1 ratio to cabergoline 0.5 mg or 1.0 mg using REDCap, irrespective of their initial treatment allocation. Study medication for the open-label phase will be supplied by the Hospital Pharmacy Clinical Trials Unit at Aarhus University Hospital.
Sample size calculation
The sample size is based on the primary endpoint, change in MMD from baseline to the end of the double-blind phase. In our pilot randomized trial, once-weekly cabergoline 0.5 mg reduced MMD by a mean of 3.6 days (SD 4.1), and no participants discontinued treatment during the 12-week treatment period. For the present trial, a conservative placebo-adjusted treatment effect of 2.8 days is assumed. With a two-sided α of 0.05 and 90% power, 47 participants per group are required. To allow for potential dropouts and to support secondary analyses, 150 participants will be enrolled.
Data collection and management
Trial data will be collected and managed using REDCap hosted at Aarhus University,35,36 with built-in validation to minimize entry errors. Data will be stored securely in compliance with GDPR, and only de-identified data will be used for analysis with access restricted to study investigators.
Statistics
The primary analyses will be based on the double-blind treatment phase, including all three randomized groups. Open-label data will solely be evaluated descriptively without formal statistical analyses.
Baseline MMD will be calculated from the 28-day prospective headache diary and considered evaluable for eligibility if at least 26 of 28 diary days are completed (≤10% missing). When 26–27 diary days are available, MMD will be prorated to a 28-day equivalent as (number of migraine days/number of observed diary days) × 28.
The primary outcome will be analysed using crude and adjusted linear mixed-effects models fitted by restricted maximum-likelihood estimation. Adjustment will be made for age, sex, baseline MMD, current use of preventive migraine medication and previous preventive treatment failure. The primary analysis will follow the intention-to-treat principle, including all randomized participants in their allocated groups. Missing outcome data will be handled within the mixed-model framework under the missing-at-random assumption, without any imputation of missing data. Sensitivity analyses using multiple imputation will be performed.
Secondary outcomes will be analysed using similar models, adjusted for the baseline value of the respective outcome, baseline MMD, age, sex and preventive migraine medication status. Binary outcomes, including the ≥50% responder rate, will be analysed using generalized linear models with a binomial distribution to estimate prevalence ratios and absolute differences. Ordinal outcomes will be analysed using ordinal logistic regression, with crude and adjusted estimates.
Additional analyses will include per-protocol analyses, subgroup analyses by sex and dose–response analyses. For dose–response analyses, treatment will be modelled as a numeric variable reflecting dose level (coded 0, 0.5 and 1.0 mg) to formally assess a linear dose–response relationship (P for trend). In addition, prespecified pairwise comparisons (0.5 mg vs placebo and 1.0 mg vs placebo) will be performed. A pooled comparison of active treatment versus placebo will be conducted. Safety outcomes will be compared descriptively between groups.
A full Statistical Analysis Plan will be finalized prior to database lock and unblinding.
Data monitoring and trial oversight
The trial is investigator-initiated and coordinated at the Department of Internal Medicine and Endocrinology, Aarhus University Hospital, with the primary investigator responsible for overall trial conduct. Data collection and management are performed by the investigative team using REDCap.
An independent Data Monitoring Committee (DMC), with expertise in clinical pharmacology, neurology and trial methodology, oversees participant safety and trial conduct. The DMC reports its recommendations to the sponsor and primary investigator. The DMC will perform an interim safety review after 30 participants have completed the double-blind phase. No interim efficacy analyses or stopping guidelines are planned. Only the DMC will have access to interim safety data, and any decision to terminate the trial will be made by the sponsor in consultation with the DMC.
Trial monitoring will be performed by an independent monitor from the regional GCP unit in accordance with Good Clinical Practice.
Current trial status
The approved protocol is Version 2.0 (26 September 2025). Recruitment began in November 2025, and study completion is planned for February 2028.
Patient and public involvement
A group of individuals with migraine and the chairperson of the Danish Patient Association for Headache Disorders (Danmarks Patientforening for Hovedpineramte) were consulted during the design phase. Their input informed the structure and content of the electronic headache diary and led to revisions of participant-facing materials to improve clarity and usability.
Discussion
Dopamine agonists improve headache in patients with hyperprolactinaemia,18–22 and in a recent pilot trial, once-weekly cabergoline 0.5 mg reduced MMD and was associated with a patient-reported overall improvement, without serious adverse events, in participants with up to 14 MMD. The effect size was comparable to established preventives, including anti-CGRP therapies, with the advantages of oral administration, favourable tolerability and low cost. 25
Despite recent therapeutic advances, an unmet need for effective and well-tolerated preventive treatments remains. 37 Moreover, the cost of migraine drugs has increased substantially with the introduction of CGRP-targeted therapies. 38 In this context, repurposing cabergoline may represent a cost-effective addition to migraine prevention and broaden access in healthcare systems where high drug costs limit treatment availability.
The present trial extends our previous work by enrolling a larger sample, testing two cabergoline doses, and including an open-label extension to evaluate persistence of effects and tolerability. In addition, the study may advance understanding of prolactin and dopamine signalling in migraine pathophysiology, informing future therapeutic strategies.
Clinical implications
Cabergoline may represent a novel, low-cost oral preventive option for migraine. The randomized, double-blind, placebo-controlled design follows current migraine treatment trial guidelines, and the use of electronic daily headache diaries enhances data quality by minimizing recall bias. Evaluation of two cabergoline doses and a 12-week open-label extension will provide information on dose–response relationships, tolerability and safety.
Footnotes
Acknowledgements
The authors thank patient contributors and the chairperson of the Danish Patient Association for Headache Disorders for their input to the trial design. ChatGPT (OpenAI) was used for language editing.
Ethical considerations
The trial has been approved by the relevant ethics committee and regulatory authorities via the European Medicines Agency Clinical Trials Information System (EU trial number: 2025-522323-10).
Consent to participate
Written informed consent is obtained by trained investigators prior to enrolment. No additional consent is required, as biological samples will not be used beyond the protocol. Any important protocol amendments will be submitted for approval and updated in applicable trial registries. Personal data will be handled in accordance with GDPR. Compensation for study-related harm will be provided under the Danish Patient Compensation system. Results will be reported in trial registries and peer-reviewed journals, presented at scientific meetings, and shared with participants in plain-language summaries.
Consent for publishing
All authors agree that, if the manuscript is accepted, it may be published in Cephalalgia Reports.
Author contributions
JOLJ and AJH conceived the study. AJH is the primary investigator and, together with RMHT, is responsible for trial conduct. KL developed the statistical analysis plan. HCD and HK provided neurological supervision. JOLJ obtained funding and serves as trial sponsor. AJH drafted the manuscript, and all authors reviewed and approved the final version.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Danmarks Frie Forskningsfond (grant number 10.46540/4308-00036B).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Data availability statement
The full study protocol and the statistical analysis plan will be available from the corresponding author on reasonable request. De-identified individual participant data and statistical code will be available from the corresponding author upon reasonable request.
Sponsor
The trial sponsor is Aarhus University Hospital, Denmark. Professor Jens Otto Lunde Jørgensen acts as the sponsor representative.
Trial registry
The trial is approved by the Danish Health Authorities and the National Committee on Health Research Ethics and registered at ClinicalTrials.gov (NCT07072910) and the EU Clinical Trials Information System (EudraCT 2025-522323-10).