
Abstract
Alzheimer's disease (AD) is a complex neurodegenerative disorder and the major cause of dementia. Amyloid-β (Aβ) and tau aggregation, mitochondrial dysfunction, and microglial dysregulation are key contributors to AD pathogenesis. Impairments in the blood-brain barrier have unveiled the contribution of the immune system, particularly B cells, in AD pathology. B cells, a crucial component of adaptive immunity, exhibit diverse functions, including antigen presentation and antibody production. While their role in neuroinflammatory disorders has been well-documented, their specific function in AD lacks adequate data. This review examines the dual role of the B cells and humoral immunity in modulating brain inflammation in AD and explores recent advancements in passive and active immunotherapeutic strategies targeting AD pathobiology. We summarize preclinical and clinical studies investigating B cell frequency, altered antibody levels, and their implications in neuroinflammation and immunotherapy. Notably, B cells demonstrate protective and pathological roles in AD, influencing neurodegeneration through antibody-mediated clearance of toxic aggregates and inflammatory activation inflammation. Passive immunotherapies targeting Aβ have shown potential in reducing amyloid plaques, while active immunotherapies are emerging as promising strategies, requiring further validation. Understanding the interplay between B cells, humoral immunity, microglia, and mitochondrial dysfunction is critical to unraveling AD pathogenesis. Their dual nature in disease progression underscores the need for precise therapeutic interventions to optimize immunotherapy outcomes and mitigate neuroinflammation effectively.
Introduction
In 1907, Alois Alzheimer, a German psychiatrist and neuropathologist, introduced a 51-year-old woman exhibiting memory dysfunctions, confusion, and disorientation in a case report. 1 Alzheimer's disease (AD) has been considered one of the most important neurodegenerative disorders. The pathobiology of AD is defined by the aggregation of amyloid-β (Aβ) and tau phosphorylation or aggregation, which disrupts neuronal functions, 2 impairs the blood-brain barrier (BBB), and recruits peripheral immune cells into the central nervous system (CNS). 3 The brain's immune privilege, maintained by the BBB and the absence of lymphatic vessels and antigen-presenting cells (APCs), is compromised in AD. 4 This impairment allows peripheral immune cells to infiltrate the CNS and interact with self-antigens, altering the brain's immune environment. 3
Accordingly, the levels of brain-infiltrated T cells and B cells are elevated in the parenchyma, perivascular and meningeal spaces, and cerebrospinal fluid (CSF) of individuals with neurological disorders and neuroinflammatory conditions, highlighting the roles of adaptive immunity in the AD pathogenesis. 5 Due to higher rates of T cell infiltration in the brain compared with B cells, studies have primarily focused on elucidating T cell roles in AD pathology. In contrast, the functions of B cells remain relatively unexplored.5,6
In addition to serving as a professional APC and a source of cytokine production, B cells differentiate into plasma cells, which produce antibodies, thereby contributing to various humoral and cellular activities. 7 B cell-derived antibodies could target pathologically accumulated Aβ and tau, facilitating their antibody-dependent clearance mediated by microglia and astrocytes. On the other hand, these antibodies may also play pathological roles by impairing the BBB, including cerebral amyloid angiopathy (CAA), and promoting neuronal damage. 8
Therefore, the current study aims to provide an overview of the interplay between innate and adaptive immunity in AD, with a particular focus on the role of B cells and humoral immunity in either the regression or progression of the disease. Besides, recent therapeutic approaches, emphasizing passive and active immunotherapy targeting B cell functions in AD, are discussed.
Alzheimer's disease
AD is considered one of the most lethal and burdening neurodegenerative disorders, primarily affecting the elderly population worldwide. While memory loss and short-term memory issues are leading signs of AD, other hallmarks include executive dysfunction, visual-spatial impairment, and neuropsychiatric disturbances. 9 The variability in cognitive impairment among patients often begins with mild cognitive impairment (MCI), characterized by single or multiple cognitive domain impairments while maintaining functional capacities. 10 Progression to dementia, marked by widespread cognitive and neurobehavioral decline, leads to significant life alternations and eventual loss of independence. 11
Epidemiological data demonstrate that AD prevalence is increasing globally, with projections estimating 113 million cases by 2050, 12 particularly in middle- and low-income countries. 13 Age is the primary risk factor, and incidence is notably higher in women, potentially due to the differences in tau pathology.14–17 Genetic factors, particularly APOE ε4 allele, significantly elevate AD risk, alongside other variants TREM2 and MAPT, though to a lesser extent.18–20
Pathophysiology of AD, accumulation of Aβ and tau proteins
AD pathology involves excitotoxic synapse loss and neuronal death, primarily driven by the aggregation of Aβ and tau proteins. These proteins disrupt synaptic transmission and cellular homeostasis, triggering immune responses that contribute to disease progression.21–24
The initial deposition of extracellular neurotic Aβ plaques in the medial temporal lobe, followed by their spread throughout the cortical regions, including the isocortical areas of the temporal, frontal, and parietal lobes.25,26 Aβ oligomers, particularly Aβ42, are highly neurotoxic and interact with receptors such as NMDA and AMPA. This interaction leads to excessive glutamate release and overactivation of NMDA receptors, causing calcium overload in neurons. The resulting excitotoxicity activates proteases like calpains and caspases, degrades synaptic proteins, destabilizes the cytoskeleton, and induces synaptic degeneration and neuronal death.26–30 In addition to receptor dysfunction, Aβ oligomers impair long-term potentiation, a process crucial for synaptic plasticity and memory formation. This further weakens synaptic connections, particularly in the hippocampus, a region for cortical memory.25,31
Tau protein, related to microtubule stabilization, undergoes post-translational modifications leading to its aggregation. These aggregates, found in the intracellular space as neurofibrillary tangles (NFTs) and in neuropil as dystrophic neurites, disrupt axonal transport and synaptic dysfunction.32,33 Tau pathology spreads trans-synaptically across connected neurons, further exacerbating synapse and neuronal loss.21,25–27 This mislocalization and aggregation have been observed in regions such as the medial temporal lobe, that is associated with cognition, 34 and in the brainstem nuclei, even in younger individuals. 35
Mitochondrial dysfunction in AD, although significant, is beyond the direct scope of this review. Both Aβ and tau affect ATP production, leading to synaptic energy deficits and contributing oxidative stress. The indirectly influences synaptic degeneration and neuronal death. 25
The immune system is a critical driver of synaptic loss in AD. Microglia, the brain's resident immune cells, are activated in response to Aβ plaques and tau aggregates. Initially attempting to clear these pathological proteins, microglia transition into a neurotoxic state over time, releasing pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-1β. These cytokines upregulate the complement system, tagging synapses with proteins like C1q and C3 for elimination. This results in excessive pruning of excitatory and inhibitory synapses, particularly around Aβ plaques.30,36 Astrocytes further contribute to this process by engulfing synaptic terminals, especially in later disease stages, exacerbating synaptic and neuronal loss. 30
Proteomic analyses of AD brains have revealed reduced levels of synaptic proteins, such as synaptophysin and PSD95, correlating with cognitive decline. This imbalance between excitatory and inhibitory synapses contributes to neuronal hyperexcitability, amplifying AD pathology. 36
The APOE ε4 allele, a major genetic risk factor for AD, further complicates the immune landscape. It influences Aβ aggregation and clearance, exacerbating the development of CAA and impairing microglial phagocytic activity.11,25,37 Carriers of the APOE ε4 allele exhibit earlier onset and greater Aβ accumulation compared to non-carriers, as shown in studies utilizing positron emission tomography (PET) imaging.38,39
The dysregulation of immune responses plays a significant role in AD pathology. Peripheral immune cells, including monocytes, neutrophils, T cells, and B cells, infiltrate the brain when the BBB is compromised. The detection of antibodies against Aβ and the presence of T cells in CSF of AD patients underscores the involvement of adaptive immunity.40–43 Additionally, evidence of autoimmune responses, such as the activation of peripheral B cells and the production of autoantibodies, suggests that humoral immunity may both contribute to and counteract disease progression.7,44
The roles of the immune system in AD
The immune system's involvement in AD was first described by Alois Alzheimer in 1907, with observation of glial cell morphological changes in patient brain samples. Since then, clinical studies and genome-wide association studies (GWAS) have demonstrated dysregulation of the immune system as a significant contributor to AD pathogenesis. 45 Chronic low-grade inflammation, often arising from aging, metabolic diseases, or pathogen exposure, has been increasingly recognized as a driver of AD progression. Elevated levels of inflammatory mediators, such as C-reactive protein (CRP), are strongly associated with higher AD risk, especially APOE4 carriers. These systematic inflammatory states correlate with accelerated neurodegeneration and brain atrophy in key regions like the hippocampus and temporal lobe, emphasizing their role in amplifying genetic susceptibilities to AD.46,47
Immune activation facilitates the infiltration of peripheral immune cells into the brain, a key mechanism in AD progression. During systemic inflammation, immune cells such as T cells and B cells can cross a compromised BBB. Once inside the brain, these infiltrating cells interact with microglia and astrocytes, amplifying neuroinflammatory responses and contributing to hallmark AD pathologies such as Aβ plaques and tau tangles. 48 Chronic inflammation shifts microglia, the brain's primary immune cells, from a homeostatic state to a pro-inflammatory phenotype, impairing their ability to clear Aβ and release neurotoxic cytokines like TNF-α and IL-1β. These cytokines exacerbate oxidative stress and synaptic dysfunction, leading to cognitive decline.49–51
Pathogen exposure further exacerbates systemic immune activation, triggering a cascade of events that impact the brain's immune environment. Infections induce trafficking of immune cells, including B cells, into the CNS, where they release pro-inflammatory cytokines and disrupt immune balance. This pro-inflammatory state impairs microglial Aβ clearance and promotes tau hyperphosphorylation, two central processes in AD pathology. This feedback loop between peripheral immune activation and neuroinflammation perpetuates AD progression.49,50
Importantly, B cells play a dual role in AD. While their antibody production targets pathological aggregates like Aβ and tau, aiding clearance through microglia activation, their dysregulation can exacerbate inflammatory processes and contribute to vascular damage, such as CAA. These findings emphasize the need to further investigate the specific contributions of B cells and their antibodies in AD to delineate their protective versus pathological roles.
Innate immune system and AD
The innate immune system is one of the two chief arms of the immune system. This innate mechanism can react the pathogens based on receptors that are encoded by the germline. 52 The characteristics of innate immunity let this system respond more rapidly but in a non-specific manner. 53 These receptors or so-called pattern-recognition receptors interact with damage-associated molecular patterns (DAMP) such as denatured DNA and/or misfolded/aggregated proteins as well as pathogen-associated molecular patterns (PAMP) like the nucleic acids, glycoproteins, and lipoproteins of pathogens which leads to the activation of intracellular signaling (NF-κB pathway) and inflammation as the main consequence of innate immune responses. 52 Dendritic cells, monocytes, neutrophils, macrophages, and natural killer (NK) cells are cell compartments involved in the innate immune arm, and the complement system is the soluble molecular compartment. 53 The mentioned components are of the peripheral immune system based on CNS; nonetheless, there is a central immune system in the CNS, including cells such as microglia and astrocytes. 54
Central immune system
Microglia
Microglia are the primary innate immune cells in the CNS 55 and play crucial roles in maintaining brain homeostasis and responding to neuroinflammatory conditions, particularly in AD. 56 Microglia, the resident immune cells of the CNS, are pivotal in the brain's defense mechanisms against pathogens and cellular debris. They exhibit a dual role, functioning as both protectors and potential contributors to neurodegeneration.57,58 In the context of AD, microglia can be categorized into various phenotypes, including disease-associated microglia, which adapt their transcriptional profiles in response to pathological conditions, such as the accumulation of Aβ plaques.59–62
The presence of Aβ plaques triggers microglial activation, leading to the secretion of pro-inflammatory cytokines such as TNF-α and IL-1β, which further exacerbate neuroinflammation. 63 Interestingly, microglia can also facilitate the clearance of Aβ through phagocytosis and the secretion of proteolytic enzymes, such as matrix metalloproteinases. 64 However, chronic activation of microglia can result in a detrimental feedback loop, where prolonged inflammation contributes to increased Aβ aggregation and neuronal death. 65 This highlights the complexity of microglial functions, where their protective roles can become maladaptive in the context of chronic neurodegeneration. 66
Astrocytes
Astrocytes also play dual roles in AD, contributing to both neuroprotection and neurodegeneration. Astrocytes maintain CNS homeostasis by regulating oxidative stress, synaptic remodeling, and BBB permeability.67–70 In AD, astrocytes are able to clear Aβ plaques through matrix metalloproteinase production.68,69,71 However, astrocytes are also involved in neuroinflammation, producing cytokines like IL-1, IL-6, and TNF- α upon to recruitment to Aβ plaques, leading to neuronal damage.72–75
Astrocytes interact with Aβ via receptors such as RAGE and LRPs, promoting inflammatory pathways and synaptic dysfunction.76–78 Impaired astrocytic phagocytosis exacerbates Aβ accumulation and neuronal apoptosis. 79 Additionally, astrocyte activation increases nerve growth factor (NGF), contributing to tau hyperphosphorylation and neurodegeneration.80,81 All in all, astrocytes exhibit protective functions, their inflammatory responses often dominate in AD pathology.
Peripheral immune system
The BBB is the description of sealed continuous endothelial cells of brain microvessels which are covered pericytes and astrocyte end-feet. 82 This special structure can limit the translocation of blood-circulating cells to the brain parenchyma by providing low paracellular and transcellular permeability and high trans-endothelial electrical resistance. However, the dysfunction of BBB in neurodegenerative disorders, including AD has been reported in neuroimaging studies. 83 Regarding the functions of the BBB, the brain is an immune-privileged organ which means that outward immune cells could not be allowed to present in the brain parenchyma. The disruption of BBB is the crucial trigger of peripheral immune cells entering the brain area. 84 Although several studies did not find any significant association between AD development and peripheral inflammation,85,86 many systematic reviews and meta-analyses demonstrated that there is evidence of dysregulated inflammatory responses in AD pathology87,88 and that there could be a relationship between peripheral inflammatory markers and higher risks of dementia.89,90
Inflammatory cytokines
Initial studies demonstrated that there is an association between the development of dementia and AD and the levels of inflammatory cytokines. Elevated levels of TNF, IL-1β, IL-6, and CRP in midlife are linked to an increased risk of cognitive decline and earlier AD onset, particularly in APOE ε4 carriers.91–95 Some inflammatory mediators peak in early AD stages and decline in advanced dementia.96–103
Specific cytokines contribute to AD pathology. Increased levels of TNF-α, IL-1β, TGF-β, IL-12, and IL-18 are observed in AD patients, while only TGF-β shows elevation in CSF. 88 IFN-γ and TNF-α promote microglia recruitment to Aβ plaques, enhancing neuroinflammation and inhibiting Aβ clearance via β-site APP-cleaving enzyme (BACE-1).104–106
Cytokines’ roles have remained complex. IL-18 enhances AβPP processing and Aβ40 production, 107 whereas IL-1β overexpression reduces Aβ plaques and increases tau phosphorylation and microglia activation. 108 IL-1β also induces S100B secretion from astrocytes, 109 leading to Aβ deposition, Aβ aggregation, and inducing cerebral amyloidosis and gliosis.110,111 Conversely, IL-33 appears protective, as its reduced expression in AD correlates with higher Aβ deposition, while overexpression Aβ40 production. 112
Immune cells
The breakdown of the BBB, observed in various neurodegenerative disorders including AD, facilitates the infiltration of peripheral immune cells into the CNS, allowing immune system recognition of brain antigens.113,114
Monocytes are recruited into the CNS through CCL2-CCR2 signaling, 115 and studies in transgenic AD models indicate that CCR2 deficiency impairs monocyte infiltration and enhances Aβ clearance, suggesting their dual role in AD pathology. 116 Additionally, TREM2+CD11b+ monocytes, distinguishable from resident microglia,117,118 accumulate around Aβ plaques, and their phenotypic shift from anti-inflammatory monocytes to proinflammatory monocytes as AD progresses correlates with altered CRP, C1s, and C8a levels. 119 Monocyte-derived macrophages, particularly in later disease stages, exhibit high TREM2, CD36, and macrophage scavenger receptor (MSR-1) expression, indicating their strong phagocytic ability against Aβ. 120
Neutrophils also contribute to AD pathology, showing increased frequency and elevated neutrophil-specific proteins (e.g., myeloperoxidase, neutrophil gelatinase-associated lipocalin) in AD patients. 121 Their recruitment into Aβ -rich brain regions is mediated by LFA-1 integrin, where they secrete IL-17 and neutrophil extracellular traps, causing neurotoxicity and BBB disruption.122–125 Interestingly, LFA-1 deficiency protects against memory loss and neuroinflammation. 122 Neutrophils also amplify microglial overactivation by releasing IL-2 and macrophage inflammatory protein, further exacerbating neuroinflammation. 126
NK cells undergo functional alternations in AD, though their frequency remains unchanged. In transgenic AD models, microglial activation triggers a positive feedback loop; overactivation NK cells and NK cell depletion reduce neuroinflammation while improving neurogenesis and cognitive function. 127 However, evidence regarding NK cells in AD remains limited, requiring further investigation. 128
Adaptive immune system and AD
The adaptive immune system, composed of T and B cells, plays a crucial role in immune surveillance, mediated by highly specific T cell receptors and B cell receptors (BCRs) generated through somatic recombination. 53 In AD, the breakdown of BBB allows peripheral immune cells to infiltrate the CNS, altering conventional immune responses. 3 These infiltrating adaptive immune cells interact with neurons, microglia, astrocytes, and oligodendrocytes, 129 influencing neuroinflammation and disease progression. 130
While B cells are more unexplored, T cell infiltration in AD-affected regions has been widely reported. CD3+ T cells, primarily CD8+, are found in the hippocampus, cortex, and perivascular spaces, correlating with tau pathology rather than Aβ plaques.131,132 Chemokines like CCL3 and CXCL10 drive their recruitment, contributing to prolonged neuroinflammation.133–136 However, T cells show contradictory roles; Th1/Th17 cells exacerbate Aβ accumulation and inflammation, whereas Tregs display both protective and harmful effects depending on disease stage.137–140
The exposure of CNS antigens due to meningeal lymphatic dysfunction suggests a potential autoimmune component in AD.141–143 Increased brain-infiltrating antibodies and auto-antibody-positive neurons suggest immune-driven neurodegeneration, possibly through complement activation.144–147 In addition, autoimmunity-related genes such as CR1 (complement receptor 1), CLU (clusterin), and BIN1 (bridging integrator) are linked to AD pathogenesis via large-scale GWAS studies, indicating an overlap between immune dysregulation and neurodegeneration.148–150
Experimental models highlight the complex role of adaptive immunity in AD. Mice lacking T and B cells exhibited reduced Aβ burden, suggesting a detrimental role for adaptive immune activation. 151 However, broader immunodeficiency (including NK cells depletion) worsened Aβ accumulation, impairing clearance mechanisms. 152
B cells and humoral immunity
B cells, along with T cells, are the main parts of adaptive immunity. The generation of antibodies upon encountering antigens is likely the most notable function of B cells. Although B cells are known for their outstanding roles in antibody production, these lymphocytes take part in many other antibody-dependent and non-antibody-dependent aspects of adaptive immunity.153–155
General concepts about B cells
Maturation and development
Initial development of B cells occurs in the fetal liver and after that, it continues in postnatal BM, a place for receiving numerous developmental signals to differentiate. 153 The early steps of B cell development are carried out via the ordered rearrangement of immunoglobulin (Ig) heavy (H) and Light (L) chain loci. 156 Hematopoietic stem cells can produce progenitor, precursor, immature (transitional), and afterward mature naïve B cells. 157 Progenitor (pro)-B cells do not express surface Ig; however, the variable (V), diversity (D), and joining (J) genes rearrangement of the H chain occurs in this stage. 158 By contrast, early precursor (pre)-B cells express μ H chain which together with surrogate L chains makes pre-BCR able to interact with ligands. 159 Indeed, the discovery of surrogate L chains was the main step in defining how early B cells could be developed. 160 Despite the previous studies,161,162 the signaling of pre-BCR could be a ligand-independent mechanism according to crystal structure solution assessing human pre-BCR. 163 With this regard, if pre-BCR cannot be activated, the B cells do not receive survival signals.153,164
BAFF (B cell activating factor), primarily known for its role in B cell survival, has gained attention for its involvement in neuronal survival and potential implications in AD. Research by Tada et al. identified the expression of BAFF and its receptor (BAFF-R) in neurons, extending the function of BAFF beyond the immune system. This study demonstrated that BAFF-R signaling promotes the survival of neural cells, with BAFF-R deficiency accelerating neuronal degeneration in neurodegenerative models such as amyotrophic lateral sclerosis. Although the study focused on amyotrophic lateral sclerosis, the findings have significant implications for AD, given the shared characteristics of neurodegeneration across these diseases. The neuroprotective effects of BAFF-R suggest that its signaling pathway might mitigate neuronal loss in AD, where neuronal death is a hallmark of disease progression. This opens up the possibility that BAFF could be targeted therapeutically to slow neurodegeneration in AD by enhancing neuronal resilience to pathological insults.165,166
Additionally, the role of BAFF in neuroinflammation, a key factor in AD pathogenesis, has also been explored. Neuroinflammatory responses, particularly those mediated by glial cells such as microglia and astrocytes, contribute to the sustained damage seen in AD. BAFF, which is involved in the regulation of immune cells, including B cells and microglia, is thought to influence these inflammatory processes. Elevated BAFF levels in the context of neuroinflammation may reflect an attempt to counteract neuronal damage by promoting protective immune responses. However, excessive or dysregulated BAFF signaling could potentially exacerbate inflammation, contributing to a harmful cycle of neuronal damage. The dual role of BAFF in both immune regulation and neuroprotection makes it an intriguing target for therapeutic intervention in AD, where modulating neuroinflammatory responses could slow disease progression.167–170
The BCR signaling (a complex consisting of surface Ig and CD79A–CD79B) is the key phenomenon in the B cell maturation by mediating the crucial B lymphocyte education plan, positive and negative selection leading to the development of an immune-competent repertoire of B cells which could not react to self-antigens.171,172 Immature B cells are lymphocytes able to migrate from the BM into the spleen and acquire CD21, CD22, and complete IgM as the surface Ig and are.173,174 The interaction of BCR and self-antigens induces the regulatory signals in the immature B cells, which end in cell death,175,176 energy,177,178 or receptor editing.179–181 This process is called central tolerance. 182 Immature B cells carry out the developmental process by migrating from BM into the spleen as transitional B cells. 183 Finally, by recognizing a cognate antigen with the help of follicular helper T cells, the mature B cells could be activated and differentiated into effector CD38+ antibody-producing plasma cells and CD27+ memory B cells. 157 The Ig class switching is a T-cell-dependent event that lets B cells produce IgG, IgE, and IgA in addition to IgM and IgD. 184 Although in each stage, B cells express some particular molecules, typically CD19 and B220 are molecules specifically related to the B cell lineage. 185
Concerning the subsets of B cells, naïve B cells could be differentiated into three subsets, including B1 B cells, conventional B2 B cells (follicular (FO) and marginal zone (MZ) B cells), and regulatory B cells (Bregs). These subsets differentiate based on the location they are in, the ability to migrate, and the T cell-dependent or -independent way of activation. 186 In 1983, a unique subset of B cells, B1 B cell, was described by Lee Herzenberg in murine. 187 It was demonstrated that despite the origin of conventional B2 B cells, the BM, B1 B cells originated from a distinct lineage in the fetal liver. 188 Based on the expression of CD5, B1 B cells are categorized into CD5+ B1a and CD5− B1b. 189 Generally, B1 B cells are responsible for providing immunity against T cell-independent antigens majorly by producing IgM responses 186 ; however, each B1 B cell type has a distinct role. B1a B cells could respond to bacterial infections by producing natural antibodies. On the other hand, B1b B cells are able to produce antibodies against T cell-independent type 2 antigens such as polysaccharides in the infections. 190 FO and MZ B cells all are conventional B2 B cells originating from transitional B cells. Based on signals they received, the genes responsible for MZ B cell differentiation are expressed when weak BCR together with NOTCH2 signaling occurs. 191 On the other hand, strong BCR signaling results in the expression of FO B cell-induced genes. 192 The marginal zone is the area surrounding germinal centers and the home for MZ B cells. They are parts of innate immunity and can differentiate into short-lived plasma cells independent of BCR ligation. They could circulate in the blood and mediate the immunity against T cell-independent antigens. 193 By transporting antigens into the splenic follicles where FO B cells are present, MZ B cells could also aid the T cell-dependent responses against pathogen proteins and lipids. 194 FO B cells are the chief subset of B cells. They reside in the follicular niche where they could activate T cells and be activated in a T cell-dependent manner against T cell-dependent antigens, i.e., proteins. 186 Initial T cell-dependent responses lead to the differentiation of FO B cells into short-lived plasma cells which are not capable of migrating but could generate antigen-specific germ-line-encoded antibodies. 153 In the following, further T cell-dependent responses stimulate the development of germinal centers in which long-lived plasma cells are generated, able to migrate to the BM and produce high numbers of antibodies. 195 Besides, memory B cells are generated following primary antibody responses and have boosted BCR to detect antigens and respond to them more rapidly. 196
Humoral immunity
B cells are responsible for humoral immunity, primarily through their production of antibodies. These antibodies recognize a wide range of antigens due to rearrangement of the variable regions of heavy (H) and light (L) chain, which confer high specificity. The effector functions of antibodies depend on their isotypes, such as IgM and IgG, which activate the complement system and mediate immune responses like antibody-dependent cellular cytotoxicity and phagocytosis. 197 In the context of AD, B cell-derived antibodies play a dual role. On one hand, antibodies target pathological aggregates like Aβ and tau, aiding in their clearance through mechanisms such as microglia activation and complement-mediated phagocytosis. On the other hand, antibody interactions can exacerbate neuroinflammation and vascular damage, including CAA, thereby contributing to AD pathology.198,199 Mature B cells, which act as APCs, amplify immune responses by presenting antigens to CD4+ T cells via major histocompatibility complex molecules. This interaction is supported by co-stimulatory signals provided by molecules like CD80, CD86, and CD40, which are critical for T cell activation. In AD, these antigen presentation processes may contribute to both protective and pathological immune responses, depending on the context.200–202 B cells also secrete cytokines that modulate adaptive immune responses. For instance, IL-4 and IL-10, secreted by regulatory B cells, exhibit anti-inflammatory effects that can mitigate AD pathology by reducing neuroinflammation and promoting Aβ clearance. However, dysregulated cytokine production, such as elevated levels of pro-inflammatory mediators like TNF-α and IL-6, may accelerate neurodegeneration and synaptic loss. 198 Variations in cytokine-related genes, such as polymorphisms in IL-4 and IL-10, have been linked to altered AD risk and progression across different populations. 199 B cells further influence CNS homeostasis through their production of lymphotoxin, which contributes to the development of secondary and tertiary lymphoid structures. These structures can modulate local immune responses, but their dysregulation in the CNS has been implicated in neuroinflammatory disorders, including AD.203,204 The role of B cells in AD highlights their dual capacity to mediate neuroprotection through antibody production and immune modulation while also contributing to neuroinflammation and disease progression when dysregulated.
B cells and AD
Despite extensive studies on other immune cells, the roles of B cells in AD pathology remain underexplored but are supported by emerging evidence. B cells, key players in humoral immunity, contribute to AD through antibody production, antigen presentation, and cytokine secretion. Their involvement in AD pathology encompasses both protective and pathological aspects.
B cell infiltration has been observed in AD brains, particularly in regions like the hippocampus and cortex. These infiltrated B cells interact with resident immune cells, potentially amplifying neuroinflammatory responses. In clinical studies, autoantibodies against brain antigens and non-brain molecules were found in the CSF and serum of AD patients, suggesting B cell activation in the disease. For instance, elevated serum levels of antibodies targeting the heavy neurofilament subunit of cholinergic neurons and anti-hippocampal antibodies have been reported, indicating humoral activity within the CNS.205,206
The antibodies produced by B cells play a dual role in AD. On the one hand, these antibodies can recognize and bind to aggregated Aβ and tau proteins, promoting their clearance through microglial activation and phagocytosis. For example, the administration of antibodies against Aβ in transgenic APP mice reduced Aβ plaque deposition and improved cognitive outcomes. Similarly, IVIg therapy in APP/PS1 mice diminished Aβ deposition, enhanced microglial activation, and shifted inflammatory responses towards M2 phenotypes, favoring plaque clearance.207,208
On the other hand, B cell-produced antibodies may exacerbate AD pathology. For instance, autoantibodies against cerebrovascular Aβ are implicated in CAA-related inflammation. The interaction of antibodies with Fc receptors expressed on microglia and neurons induces pro-inflammatory cytokine release and neuroinflammation. Elevated expression of FcγRI and FcγRIII in AD brains has been associated with inflammation mediated by ERK and NF-κB pathways. Moreover, antibodies can interact with tau proteins, contributing to tau aggregation and early neurodegeneration. 209
The depletion of B cells has provided further insights into their role in AD. Studies using anti-CD20/B220 antibodies in 3×TgAD mice demonstrated that B cell depletion significantly reduced Aβ deposition and improved spatial memory. Interestingly, this intervention also modulated microglial phenotypes, reducing neuroinflammation and shifting immune responses toward homeostasis. However, the absence of B cells may impair immune surveillance and antibody-mediated clearance mechanisms. 210
Natural autoantibodies, a subset of B cell-produced antibodies, have shown neuroprotective effects in AD. These antibodies bind to Aβ oligomers and facilitate their clearance, improving spatial memory in transgenic AD mice. However, their levels decline with aging and in neurodegenerative conditions like AD, highlighting the potential of replenishing or modulating natural autoantibodies as a therapeutic approach. 211
The aging process exacerbates B cell dysfunction in AD. Immunosenescence not only reduces naïve B cell populations but also leads to an accumulation of age-associated B cells (ABCs), a distinct subset of B cells linked to chronic inflammation and autoimmunity. ABCs are characterized by the expression of CD11c and T-bet, increased secretion of pro-inflammatory cytokines such as TNF-α, IFN-γ, and IL-6, and a heightened response to innate immune signals. 212 In AD, ABCs have been implicated in promoting neuroinflammation and impairing microglial Aβ clearance, potentially accelerating disease progression. Clinical studies have shown that age-related changes in B cell subsets correlate with increased amyloid burden, tau pathology, and cognitive decline. Furthermore, ABCs have been found to expand disproportionately in individuals with chronic inflammatory conditions, including AD, contributing to sustained immune activation. Additionally, sex differences in B cell responses influence AD progression. Female carriers of the APOE4 allele exhibit heightened ABC expansion, increased B cell activation, and antibody production, which may account for their greater susceptibility to AD compared to males. 213
FDA-approved monoclonal antibody therapies, such as aducanumab and lecanemab, further underscore the significance of B cells in AD. These therapies mimic the antibody responses of B cells, targeting aggregated Aβ for clearance while attempting to mitigate neuroinflammatory side effects. Their development highlights the therapeutic relevance of harnessing or modulating B cell activity in AD. 205
Taken together, B cells play a complex role in AD pathology, exhibiting both beneficial and detrimental effects. While their antibodies contribute to Aβ and tau clearance, they may also drive neuroinflammation and BBB disruption. Further research is essential to delineate these roles and optimize immunotherapeutic strategies targeting B cells in AD.
Table 1 provides a comprehensive summary of the roles of various immune cells the pathogenesis and progression of AD.
The role of immune cells in AD.

AD: Alzheimer's disease; Aβ: amyloid-β; BBB: blood-brain barrier; CAA: cerebral amyloid angiopathy; CNS: central nervous system; IL-1β: interleukin-1β; MMPs: matrix metalloproteinases; TNF-α: tumor necrosis factor-α.
Recent therapeutic approaches targeting AD
Although numerous aspects of AD pathology have been studied and illuminated in the last two decades, the last approved drug by the Food and Drug Administration (FDA) was memantine back in 2003. With this regard, only rivastigmine, donepezil, and galantamine, which are acetylcholinesterase enzyme inhibitors, and memantine, which is an antagonist of N-methyl-D-aspartate (NMDA) receptor, are FDA-approved. 214 As stated, AD pathobiology is tightly associated with the extracellular accumulation of Aβ plaques (Aβ pathology) and intracellular deposition of tau proteins (tauopathy). Therefore, possible therapeutic approaches could target these two hallmarks. Although targeting Aβ has shown discrepancies regarding the clinical impacts of AD cases,215,216 targeting tau, on the other hand, seems to be more promising in preclinical studies, and currently they are evaluating at the clinical level. 217
Targeting Aβ
Targeting Aβ as an AD-promoting agent could be done in different ways. Drugs could impede the accumulation of Aβ plaques, diminish the production of Aβ by interfering with producer enzymes, and enhance the process of Aβ clearance. It could be inferred that the toxicity of Aβ protein is derived from its ability to aggregate. Thus, some strategies focus on hindering the aggregation of Aβ into oligomers. Scyllo-inositol (ELND005) is one of these and was shown to be a promising drug in AD models. 218 However, the outcomes of some phase II trials did not demonstrate significant differences for the AD Cooperative Study-Activities of Daily Living (ADCS-ADL) scale, cognitive and behavioral states.219,220 Tramiprosate is another anti-aggregation agent which similar to Scylla-inositol was not able to improve the cognitive decline in mild to moderate AD cases. However, reanalyzing this drug in mild AD patients carrying the APOE ε4 allele showed clinical efficacy by improving the AD Assessment Scale-Cognitive Subscale (ADAS-Cog) and stabilizing cognition for more than 78 weeks. 221
The application of BACE-1 inhibitors could disrupt the production of Aβ. Numerous studies evaluated BACE-1 inhibitors; however, the results were not promising, particularly in phase III clinical trials. It was demonstrated that lanabecestat did not improve cognitive decline in AD cases in a phase III trial. 222 Similarly, verubecestat could not slow down the impairment of cognition and function of AD patients, while it showed a high rate of adverse events (AEs). 223 To prevent the formation of Aβ plaques before the onset of AD, the efficacy of BACE-1 inhibitors has been evaluated in patients with MCI and no AD diagnosis or in individuals with normal cognition and increased Aβ levels. In this context, verubecestat, atabecestat, elenbecestat, and umibecestat were associated with cognitive worsening in drug-receiving cases instead of cognitive improvement, ending in the discontinuation of those BACE-1 inhibitors.224,225 Similarly, γ-secretase inhibitors are able to impede the production of Aβ. Evaluating the efficacy of semagacestat (a γ-secretase inhibitor) in a phase III trial was discontinued before the trial's completion. Indeed, semagacestat not only worsened the cognition and function (in daily living) of AD patients but also induced skin cancer and weight loss. 226 Besides, avagacestat, which is a γ-secretase inhibitor, showed similar outcomes in a phase II trial, resulting in the termination of the trial because of inducing skin cancers as well as no clinical effects on cognitive decline. 212 The harmful side effects of γ-secretase inhibitors could concern the Notch signaling as Notch is another substrate of γ-secretase responsible for the regulation of cell differentiation and proliferation. 227
The other way to target Aβ is to induce the removal of Aβ proteins via immunotherapy methods. This strategy has been the most effective method to hamper the progression of AD. 214 It is worth mentioning that immunotherapeutic approaches in the AD context could be categorized into active and passive based on the application of vaccines and exogenous antibodies, respectively. Active immunotherapy has higher efficacy because of its greater capacity to induce antibody production and long-lasting immune responses. Passive immunotherapy promotes immunity quickly in elderly cases that have lower responsiveness to vaccines. 228 In active immunotherapy, the strategy is based on vaccination principles, i.e., exposing the immune system to Aβ protein or its particles, which stimulates the immune cells against this protein and, consequently, immune responses occur. 229 The initial attempts regarding vaccination in AD were back in 1999 when the induction of immune responses against Aβ decreased the accumulation of Aβ in the brains of mouse models. 230 In the following, a vaccine against Aβ, AN1792, was evaluated in clinical trials. Nonetheless, the application of AN1792 was discontinued in trials due to the induction of T cell-mediated meningoencephalitis.228–231 The development of active immunotherapy in AD cases resulted in the second generation of vaccines in which the T cell epitope was depleted; however, the presence of severe AE has always been a challenge.
As stated, AN1792 was the first vaccine against full-length Aβ42. This vaccine was not able to improve ADAS-Cog, Disability Assessment for Dementia, and clinical dementia rating. Besides, AN1792 caused meningoencephalitis in 18 out of 300 patients (6%), which was the reason for trial termination. 231 In an interesting study, 22 patients from the first AN1792 trial were followed up for a 15-year postmortem. In 9 out of 16 patients, evidence of Aβ plaque clearance was obvious (extreme or intermediate), and in 5 patients, very limited Aβ plaque clearance was reported. Also, the levels of tau were low in regions of cortical foci with removed plaques compared with regions with plaques. This study indicated that long-lasting immune responses against Aβ in AD patients resulted in Aβ plaque removal. 232 The text generation of vaccines was developed in order to reduce T cell-mediated toxicities. CAD106 is a vaccine designed to interact with N-terminus Aβ1–6 and induce antibodies against Aβ (B cell responses), not T cell-mediated anti-Aβ responses. In the phase I trial, CAD106 demonstrated no clinical meningoencephalitis, while major patients exhibited anti-Aβ antibody responses. 233 Strong anti-Aβ antibody responses (IgG) were observed in phase IIb of CAD106 with more AE incidence rate, suggesting the tolerability of CAD106 with no CNS inflammation records. 234 Nonetheless, in the newest trial of CAD106 in phase II/III, unexpected alterations in cognition and brain volume led to the termination of the trial (NCT02565511). UB-311 is another novel vaccine able to stimulate B cells and conjugated helper T cells against two synthetic Aβ1–14. Indeed, the activation of Th2 could enhance the immune responses while diminishing the T cell-mediated inflammatory reactions. This vaccine-induced anti-Aβ antibody responses in 100% of AD cases and improved ADAS-Cog in mild-to-moderate AD patients. 235 ABvac40 is an active vaccine against the C-terminal end of Aβ40 and was assessed in a phase I trial. Accordingly, no evidence of amyloid-related imaging abnormalities concerning vasogenic edema, sulcal effusions, micro hemorrhages, and hemosiderin deposits was observed. Besides, in 92% of patients, the anti-Aβ antibody responses occurred, implying a satisfying safety and tolerability profile with the capacity to induce immune responses in mild-to-moderate AD patients. 236
Passive immunotherapy is defined as the functions of anti-Aβ injected antibodies. Indeed, the principles of antibody administration originated from the nature of humoral immunity and B cell responses which can promote the phagocytosis-dependent clearance of Aβ and modify the secondary structure of Aβ monomers through an antibody-based manner to impede the oligomer formation of Aβ.237,238 The primary application of passive immunotherapy was a preclinical study in which peripheral application of anti-Aβ antibodies resulted in reduced Aβ plaque deposition in APP transgenic mice. They showed that the administered antibodies were able to penetrate CNS and target Aβ, which opsonizes those proteins for microglia. 239 Numab is one of the most important monoclonal antibodies applied in AD. Being an anti-Aβ IgG, aducanumab was shown to be able to target both soluble and insoluble forms of Aβ in patients with prodromal or mild AD; however, amyloid-related imaging abnormalities were observed. 240 Recently, the effects of aducanumab in reducing biomarkers related to AD pathology were demonstrated in a phase III study of mild AD cases (EMERGE). 241 Nonetheless, there are still some doubts about the validity of the results as the FDA Peripheral and Central Nervous System Drugs Advisory Committee published a review revealing opposite opinions about the efficacy and safety of aducanumab in the treatment of AD. 242 The first monoclonal antibodies entered in the phase III clinical trial were bapineuzumab and solanezumab. Evaluation of these drugs in patients with mild-to-moderate AD showed a lack of effectiveness in improving cognition and functional abilities. 243 Although the higher incidence of AEs and amyloid-related imaging abnormalities with effusion or edema and microhemorrhage led to the termination of bapineuzumab evaluation in clinical trials, 244 the safety evaluation of solanezumab showed a safer profile according to a lower prevalence of amyloid-related imaging abnormalities. 214 The application of crenezumab in mild-to-moderate AD cases in phase II like gantenerumab application in prodromal AD patients in a phase III trial 245 did not yield clinical effectiveness in improving the cognition scores. However, regarding the favorable safety profile of those drugs, the administration of them in higher dosages is going to be considered in future trials. As one of the recent monoclonal antibodies reaching a phase III clinical trial, lecanemab (BAN2401) exhibited a favorable safety profile in the phase I trial 246 as well as a remarkable capacity to reduce the levels of brain Aβ and slow the cognitive decline in AD cases in an ongoing phase II trial. Accordingly, lecanemab was able to decrease ADAS-Cog in a dose-dependent manner over 18 months. Also, the diminished cognitive decline was higher in the APOE ε4 allele carriers. The near 10% incidence of amyloid-related imaging abnormalities- edema/effusion indicated satisfying tolerability of lecanemab in the treatment of AD. 247 Due to promising outcomes, two phase III trials, Clarity AD (NCT03887455) and AHEAD 3–45 (NCT04468659), are underway, aiming to assess the long-term efficacy and safety of lecanemab in AD cases. According to the recently published results of NCT03887455, the application of lecanemab in AD patients with MCI or mild dementia resulted in a decrease in brain Aβ burden as well as ADAS-cog and AD Composite Score (ADCOMS). This therapy induced amyloid-related imaging abnormalities in 12.6% of patients, implying that lecanemab is well-tolerated and has efficacy in improving cognitive decline. 248
Targeting tau
As stated, hyperphosphorylated tau proteins lose their capacity to bind microtubules, leading to the formation of NFTs and aggregated tau. 249 Besides, tauopathy has been demonstrated to have a better correlation with AD pathogenesis compared with Aβ pathology, suggesting the worthiness of tau proteins as a more promising target in AD therapy.228,250 Similar to Aβ targeting methods, tau targeting strategies consist of inhibiting the hyperphosphorylation of tau proteins, their aggregation, and their clearance (immunotherapy). It has been demonstrated that glycogen synthase kinase 3-beta (GSK-3β) has a chief role in the hyperphosphorylation of tau proteins. 237 With this regard, targeting GSK-3β was shown to reduce the phosphorylation of tau proteins as well as the accumulation of Aβ and neuronal loss in preclinical studies. 238 Lithium could be utilized in AD cases concerning its ability to block GSK-3β. In a phase II trial, the application of lithium in prodromal AD cases improved cognitive decline and decreased the concentration of phosphorylated tau proteins in CSF. 239 Also, the administration of a microdose of 300 μg lithium to AD patients impeded cognitive loss, 240 implying the therapeutic capacity of lithium in AD therapy. Leuco-methylthioninium bis (hydromethanesulfonate) (LMTM) is a selective inhibitor of tau aggregation. The application of LMTM in a phase III trial in mild-to-moderate AD patients resulted in no significant changes related to ADAS-Cog and ADCS-ADL scores. However, the less than 1% incidence rate of amyloid-related imaging abnormalities suggests a favorable safety profile for LMTM. 241 In the following, reanalyzing monotherapy LMTM in mild AD patients showed that 4 mg twice a day yielded similar clinical outcomes to higher doses and resulted in a reduced brain atrophy rate and an improved ADAS-cog and ADCS-ADL. 242
Immunotherapeutic approaches (passive and active) could be utilized in order to improve the process of tau clearance by targeting abnormal aggregated tau and not the physiologic ones. 243 However, an obvious challenge is the location of aggregated tau inside the cells, which could hamper the interaction of antibodies and tau proteins. Studies revealed that antibodies can penetrate neurons majorly via receptor-mediated endocytosis phenomenon (Fc-related action) and less by bulk endocytosis. 217 To induce active immunization, synthetic peptides that could mimic the pathology of tau have been utilized. AADvac1 is an active vaccine against pathological tau and its application in mild-to-moderate AD patients resulted in the stimulation of anti-tau antibody response (IgG) in all the patients associated with lower hippocampal atrophy rate and cognitive decline. Besides, no evidence of meningoencephalitis or vasogenic edema was reported. 251 A recent phase II trial (ADAMANT) exhibited the satisfactory tolerability of AADvac1 in patients with mild AD with low AE incidence rate and also high levels of IgG antibodies. However, cognitive and functional analysis showed no significant differences upon vaccination. 244 Passive immunotherapy is mediated by antibodies able to target specific sites of tau proteins. In this context, monoclonal administered antibodies should penetrate the neurons and block tau proteins. Besides, they could target tau proteins outside the neurons and guarantee the inhibition of AD progression. 217 Accordingly, gosuranemab, which is an anti-tau monoclonal antibody, was administered in patients with progressive supranuclear palsy, a rare neurological disease that involves neuron death by the aggregation of tau proteins. In this condition, gosuranemab resulted in a 98% reduction of unbound N-terminus tau levels in the CSF and a safety profile similar to the control group. 241 However, the application of gosuranemab in a phase II trial of patients with early AD was discontinued due to drug ineffectiveness on cognitive decline and functional impairment (NCT03352557). By targeting all six human tau isoforms, semorinemab (RO705705) is an IgG4 monoclonal antibody able to block extracellular tau proteins in AD patients. Despite a favorable safety profile, semorinemab was not able to improve AD hallmarks. Also, a phase II trial demonstrated that semorinemab failed in ameliorating AD conditions. 245 Another monoclonal antibody targeting the N-terminus of tau is tilavonemab (ABBV-8E12) which was well-tolerated in phase I of AD cases. 252 Nonetheless, the evaluation of tilavonemab in the phase II trial in patients with early AD did not show promising outcomes (NCT02880956) and thus, the administration of tilavonemab in AD treatment was terminated. Zagotenemab (LY3303560) is another monoclonal antibody targeting an early pathological conformation of tau. Although the safety assessment of zagotenemab was performed in two phase I trials (NCT02754830 and NCT02754830), the recently reported outcomes of a phase II trial showed that zagotenemab missed its primary endpoint in AD treatment. 228
It could be inferred that the notion behind immunotherapy in AD came from the capacity of B cells to produce autoantibodies against Aβ proteins in cases with AD. 247 From then, studies investigated the levels of circulated autoantibodies in AD patients to illuminate the roles of B cell-produced antibodies in AD248,249 leading to the consideration of B cell-mediated humoral responses as therapeutic agents in AD treatment. It is worth mentioning that the major therapeutic approaches in the context of AD treatment were based on targeting Aβ pathogenesis over the past 25 years. 214 However, neither non-immunotherapy methods, e.g., approaches based on the inhibition of accumulation of Aβ and its generation nor immunotherapy methods consisting of active and passive immunotherapy to enhance the clearance of Aβ deposit) demonstrate satisfying outcomes in improving cognitive and functional impairment in AD cases, except for some studies. Therefore, the strategies in the AD treatment changed to the context of tauopathy targeting. In this regard, clinical trials concerning anti-tau investigation are in their initial steps. In addition to strategies such as preventing the hyperphosphorylation of tau and its aggregation, immunotherapy targeting pathologic tau has received attention recently, so no clinical trial has reached phase III. Nevertheless, ongoing trials are evaluating active immunotherapy like NCT04445831 (a phase I/II clinical trial evaluating ACI-35, an active vaccine) and passive immunotherapy such as NCT03828747 (semorinemab assessment in moderate AD), NCT04867616 (the evaluation of bepranemab in a phase II trial), NCT04619420 (a phase II study aiming to investigate the roles of JNJ-63733657 in cognitive decline in AD cases), NCT04971733 (E2814 evaluation in a phase I/II clinical trial in mild AD), and NCT04149860 (a phase I safety assessment of Lu AF87908 in AD patients). Also, the roles of IVIg in ameliorating the pathogenesis of AD have been demonstrated in preclinical studies with promising outcomes. 204 However, their application in clinical trials was not as pleasing as preclinical studies. It was shown in a phase III clinical trial that two doses of IVIg for 18 months (0.2 and 0.4 g/kg every 2 weeks) exhibited a favorable safety profile with no clinical evidence regarding AD treatment. 250
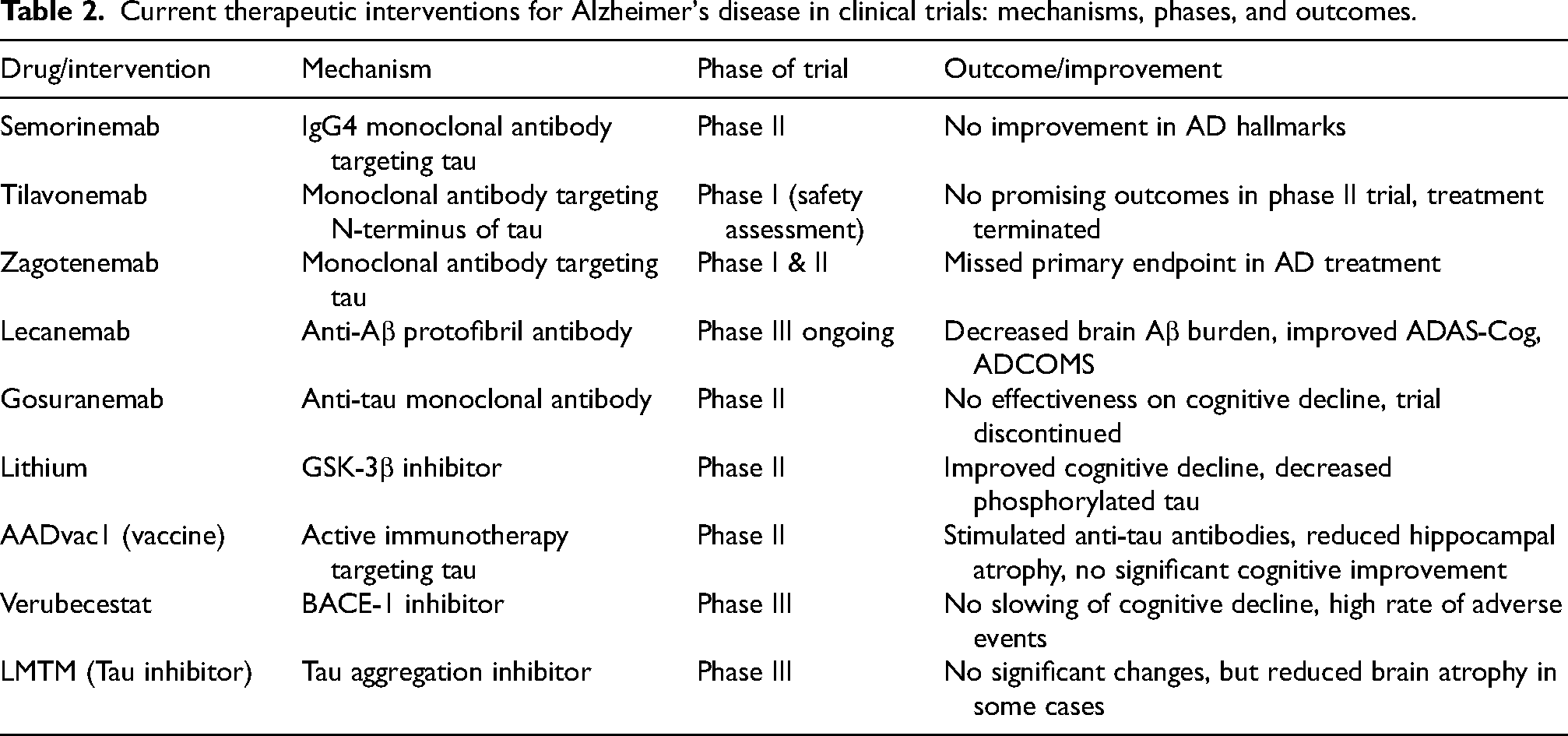
Table 2 outlines the current therapeutic interventions for AD under clinical trials, detailing their mechanisms of action, clinical phases, and reported outcomes.
Current therapeutic interventions for Alzheimer's disease in clinical trials: mechanisms, phases, and outcomes.
Conclusion
Under physiological conditions, synaptic maintenance is supported by innate immune cells such as microglia and astrocytes, which clear debris and sustain homeostasis. The brain's immune privilege, maintained by an intact BBB, CSF flow, and the meningeal and dural lymphatic systems, typically prevents the interaction of peripheral immune cells with CNS antigens. In AD, the breakdown of these protective systems disrupts CNS homeostasis. Extracellular neuritic Aβ plaques and intracellular NFTs, hallmarks of AD, persist in the brain due to the limited clearance capacity of microglia and astrocytes, leading to sustained neuroinflammatory responses and dystrophic neurites.
A dysfunctional BBB allows peripheral immune cells, including B cells, to infiltrate the brain parenchyma. This infiltration alters the immune landscape and may promote the recognition of CNS antigens as “non-self,” a hallmark of autoimmune pathology. While the presence of B cells in the brain during AD has been demonstrated, their role remains complex and controversial. B cells can produce autoantibodies targeting Aβ and tau proteins. In some studies, these autoantibodies correlate with worsened AD pathology, potentially through the promotion of proinflammatory cascades mediated by Fc receptor interactions on microglia and astrocytes. This aligns with findings that B cell depletion improves spatial memory and reduces Aβ plaque formation in preclinical models, suggesting a detrimental role for B cells in these contexts.
Conversely, B cell-derived antibodies may have protective effects. Autoantibodies against Aβ oligomers and tau tangles can facilitate their clearance through mechanisms such as antibody-dependent phagocytosis mediated by microglia. This dual role of B cells highlights their potential as both pathological mediators and therapeutic targets. The use of intravenous immunoglobulin (IVIg) and monoclonal antibodies targeting Aβ plaques, such as FDA-approved therapies Aducanumab and Lecanemab, demonstrates the therapeutic relevance of modulating B cell activity in AD. While these therapies focus primarily on Aβ clearance, emerging data suggest that targeting tau pathology might yield more favorable outcomes. B cell-derived antibodies could play a critical role in such strategies, as they can target both Aβ and tau aggregates.
Despite advancements in understanding the immune system's role in AD, significant gaps remain in elucidating the precise functions of B cells. Immunotherapy trials targeting Aβ pathology have shown mixed results, potentially reflecting the complex interplay between protective and detrimental B cell functions. For example, while active immunization strategies failed to demonstrate clear cognitive benefits, passive antibody therapies have shown promise, albeit with limitations related to side effects such as amyloid-related imaging abnormalities.
Future research should aim to clarify the conditions under which B cell activity contributes to neuroprotection versus neuroinflammation. This understanding will inform the development of more targeted immunotherapies, potentially focusing on tau pathology, given its closer association with cognitive decline in AD. By addressing the dual roles of B cells and refining strategies to harness their therapeutic potential, we may advance the efficacy of immunotherapy in AD.
Footnotes
Acknowledgments
The authors have no acknowledgments to report.
Author contributions
Ladan Gol Mohammad Pour Afrakoti (Conceptualization; Writing – original draft); Sanam Daneshpour Moghadam (Conceptualization; Data curation; Writing – original draft); Pezhman Hadinezhad, M.D (Conceptualization; Project administration).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical considerations
Not applicable.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.