
Abstract
Background
Although observational studies have reported correlations between lipid, circulating metabolites, and Alzheimer's disease (AD), the causality of these associations remain unresolved.
Objective
To investigate the effects of lipids and circulating metabolites on AD risk.
Methods
We applied a two-step Mendelian randomization (MR) framework to evaluate the causal effects of lipids on AD and to assess whether these effects are mediated by circulating metabolites. Causal estimates were primarily derived using the inverse-variance weighted method, with robustness evaluated through a series of complementary sensitivity analyses, including Cochran's Q statistic, MR-Egger intercept testing, Radial MR, MR-PRESSO, and leave-one-out analyses. Additional validation was provided by Steiger directionality testing, linkage disequilibrium score regression, multivariable MR analyses, and independent replication, collectively ensuring the reliability of the causal inferences.
Results
MR analyses showed that PI(18:1/18:1) and TAG(52:3) were causally associated with a lower risk of AD. Mediation analyses further identified triglyceride levels in low-density lipoproteins (LDL), medium LDL, and small LDL as intermediates, accounting for 17.6%, 22.8%, and 18.1% of the protective effect of PI(18:1/18:1) on AD risk, respectively. By contrast, the free cholesterol to total lipid ratio in medium high-density lipoproteins (HDL) emerged as a deleterious mediator, explaining 13.0% of the protective association between TAG(52:3) and AD.
Conclusions
This study highlights the importance of PI(18:1/18:1) and TAG(52:3) in AD development, and suggests that triglyceride levels in LDL, medium LDL, and small LDL, as well as the free cholesterol to total lipids ratio in medium HDL, may act as mediators in this pathway.
Introduction
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disorder marked by a gradual decline in cognitive function. 1 With the accelerating global aging population, the number of individuals with dementia is projected to exceed 152 million by 2050, and AD is the leading cause of dementia. 2 Although core pathological hallmarks of AD—such as amyloid-β (Aβ) deposition, tau protein hyperphosphorylation, and substantial neuronal and synaptic loss—have been identified, its exact etiology and pathogenesis remain controversial, and no effective treatments are currently available.3,4
Recent studies have suggested that AD progression is a continuous process characterized by abnormal biomarker accumulation, neuronal injury, and cognitive decline, in which lipids may play a critical role.5,6 For instance, a lipidomic analysis showed that the brain of AD patients exhibits significant lipid metabolic abnormalities compared with healthy controls. 7 Moreover, dysregulated lipid metabolism is thought to promote Aβ deposition and accelerate AD progression.8–10 Furthermore, evidence indicates that vascular dysfunction is an important component of AD pathophysiology, and alterations in circulating plasma proteins may serve as markers of vascular impairment. 11 However, disturbances in lipid metabolism can lead to alterations in multiple downstream metabolites, thereby contributing to the development of AD. 12 Multiple studies have reported that dysregulation of lipid metabolism–related pathways is accompanied by distinct changes in circulating metabolite profiles in individuals with AD relative to cognitively normal controls, and these metabolic changes are closely linked to core AD pathology. 13
However, findings from observational studies are susceptible to biases arising from confounding factors and reverse causation. As a result, the specific contributions of lipids and circulating metabolites in the onset and progression of AD need to be clarified using research approaches with stronger causal inference capability. Mendelian randomization (MR) leverages genetic variants as instrumental variables (IVs) to strengthen causal inference, thereby mitigating confounding and reverse causation that limit traditional observational analyses. 14
In this study, we used the largest currently available genome-wide association study (GWAS) datasets on lipids, circulating metabolites, and AD to conduct comprehensive MR analyses. Unlike previous studies, we not only applied univariable MR (UVMR) to estimate the overall causal effects of lipids and circulating metabolites on AD but also used multivariable MR (MVMR) to examine their independent contributions to AD risk. Our aim was to systematically elucidate the potential contributions of these factors to AD pathophysiology and to offer new theoretical insights and perspectives for AD prevention and intervention strategies.
Methods
Study design
As shown in Figure 1, we adopted a two-stage MR analytical framework to delineate the causal association between lipids and AD. We further evaluated whether circulating metabolites mediate this pathway. In the first stage (Step C), we conducted an initial assessment of the causal relationships between 179 lipid species and AD risk. To ensure robust causal inference, we applied the Steiger directionality test and bidirectional MR to verify causal direction, performed linkage disequilibrium score regression (LDSC) to examine genetic correlation and sample overlap, and used MVMR to adjust for potential confounders. Moreover, significant associations identified at this stage were further subjected to replication analyses in independent cohorts and validated through meta-analysis. In the second stage (Steps A, B, and C’), the study focused on the potential mediating roles of circulating metabolites within the established “lipid-AD” pathway. All genetic variants used as IVs satisfied the three core assumptions of MR: relevance, independence, and exclusion restriction. 15 As only publicly available GWAS summary statistics were analyzed, no additional ethical approval was required. The study was conducted in accordance with the STROBE-MR guidelines. 16
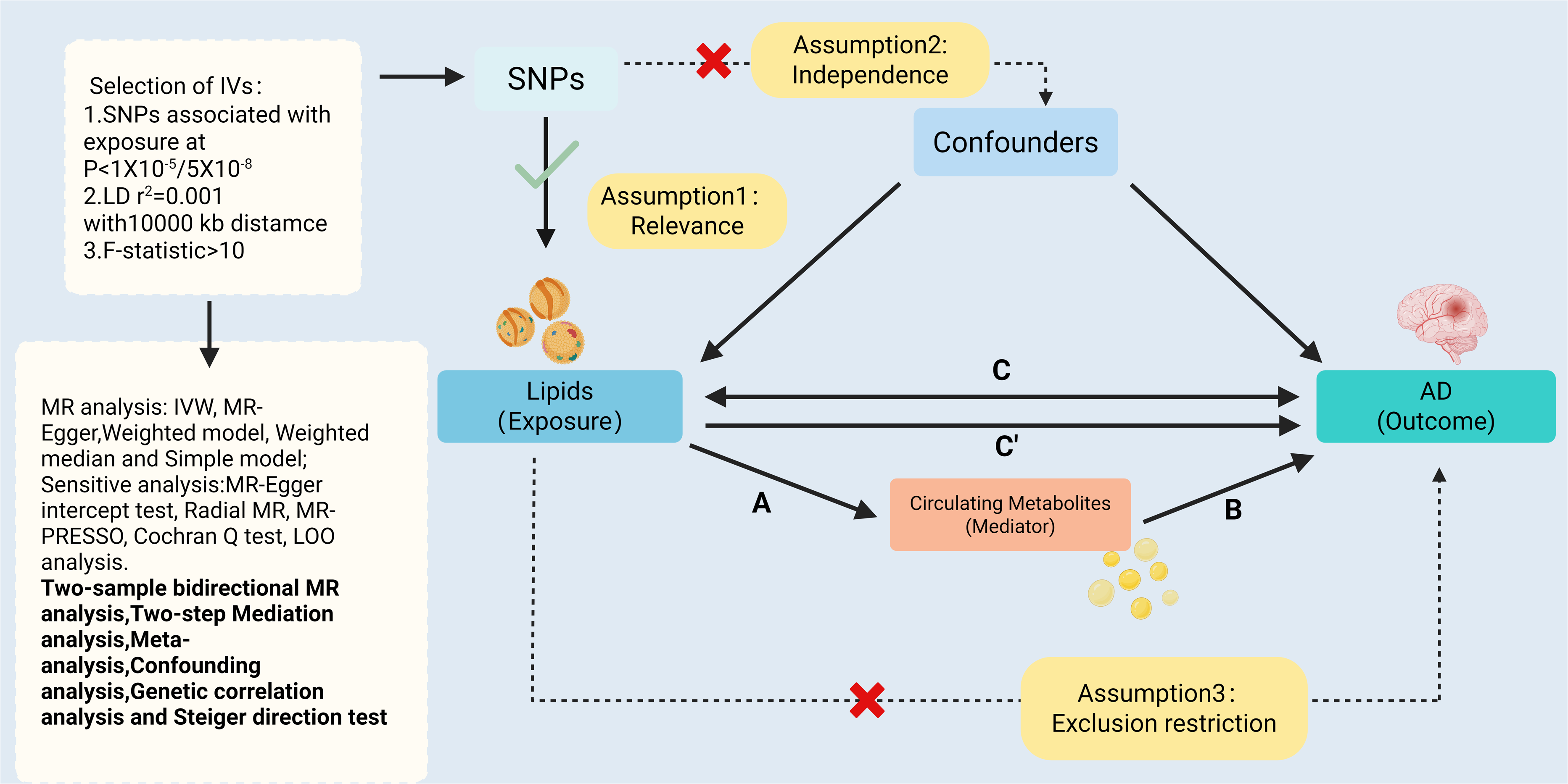
Overview of the Mendelian randomization study design.
Data sources
GWAS data for lipid phenotypes were obtained from the GeneRISK cohort, which performed shotgun lipidomic profiling on 7174 Finnish participants and identified 179 lipid phenotypes. Summary statistics were accessed via the GWAS Catalog (GCST90277238–GCST90277416). 17 The AD GWAS data were derived from Phase 1 summary statistics of the European Alzheimer & Dementia Biobank (EADB), comprising 85,934 European-ancestry cases and 401,577 controls. 18 For replication, we used the combined dataset from the International Genomics of Alzheimer's Project (IGAP) and the UK Biobank, which included 75,024 AD cases and 397,844 controls. 19 GWAS summary statistics for circulating metabolites were obtained from a large-scale meta-analysis encompassing 136,016 participants, covering 233 metabolite phenotypes, and were retrieved from the GWAS Catalog (GCST90301941–GCST90302173). 20 To minimize potential bias arising from sample overlap, all GWAS datasets were sourced from independent international consortia. Detailed descriptions of each dataset are provided in Supplemental Table 1.
Selection of instrumental variables
In this study, single nucleotide polymorphisms (SNPs) were defined as IVs. To ensure the robustness of the MR analyses, we applied a stringent quality control procedure, beginning with the selection of SNPs associated with the exposure at genome-wide significance (p < 5 × 10−8). However, the number of lipid-related IVs was insufficient under this threshold. Therefore, we relaxed the criterion to p < 1 × 10−5, consistent with previous MR studies, 21 while retaining p < 5 × 10−8 for AD and circulating metabolites. Next, to ensure IV independence, we performed linkage disequilibrium (LD) clumping using thresholds of r2 < 0.001 and a 10,000 kb window to remove highly correlated SNPs. To minimize bias from weak instruments, we calculated the F-statistic for each SNP and removed those with F-statistics < 10. 22 Finally, during harmonization, SNPs with allele mismatches or palindromic structures were removed to avoid potential bias arising from strand ambiguity or allele misalignment.
Primary MR analysis
To evaluate the causal effects of lipids on AD, we conducted two-sample MR analyses. The causal estimates were primarily derived using the inverse-variance weighted (IVW) method and were complemented by four additional approaches: weighted median, MR-Egger regression, simple mode, and weighted mode. Under the assumption that all IVs are valid, the IVW approach evaluates the overall causal effect through a weighted regression of SNP-specific Wald ratios. 23 The weighted median method yields consistent estimates even when up to 50% of the IVs are invalid, 24 whereas MR-Egger regression can provide a valid causal estimate even when all IVs exhibit pleiotropy. 25 The simple mode and weighted mode methods generate robust causal estimates when the majority (or the most heavily weighted) IVs are valid. 26 Results were considered statistically significant only when the IVW p-value was <0.05 and the direction of β was consistent across all five methods. To account for multiple testing, Bonferroni correction was applied to the IVW results, resulting in a significance threshold of p < 2.79 × 10−4 (0.05/179). 27
Sensitivity analyses
For sensitivity analyses, we used Cochran's Q test and the MR-Egger intercept test to assess heterogeneity and horizontal pleiotropy, with p > 0.05 indicating no substantial evidence of either. Outlier detection and correction were further performed using MR-PRESSO and Radial MR to improve the robustness of causal estimates.28,29 Additionally, leave-one-out analyses were applied by iteratively excluding each instrumental variable to assess the influence of individual genetic variants on the overall results. 30
Reverse causality assessment
To minimize potential bias from reverse causation, we conducted reverse MR analyses along with the Steiger directionality test. The Steiger directionality test compares the variance explained by IVs in the exposure versus the outcome to determine the direction of causality, thereby validating the consistency of the causal inference. 31
Genetic correlation analysis
To prevent bias in causal estimates arising from potential genetic overlap between the exposure and outcome, we assessed their genetic correlation using LDSC analysis. 32 The significance threshold for the genetic correlation coefficient (rg) was set at p < 0.05; p > 0.05 indicated no significant genetic correlation between the exposure and outcome, whereas p < 0.05 suggested that the causal effect may be influenced by genetic correlation or sample overlap.
Confounder analysis
To further delineate the true causal effects between lipids and AD, we conducted MVMR analyses. This approach enables the simultaneous adjustment for multiple potential confounders, thereby allowing estimation of the direct effect of the exposure on the outcome. 33 The confounders included in this study were alcohol consumption, physical activity, and sedentary behavior, all of which have been implicated as potential contributors to AD risk.34,35 Detailed sources of these data are provided in Supplemental Table 1.
Meta-analysis
To ensure the reliability of the findings, lipid phenotypes that showed significant associations in the primary MR were evaluated in independent AD summary datasets during the replication stage. We then performed a meta-analysis of the IVW estimates from the discovery and replication analyses. Statistical significance in the meta-analysis was defined as p < 0.05.
Mediation analysis
To further clarify the mechanisms through which lipids influence AD, we applied a two-step MR approach to evaluate the mediating role of circulating metabolites in the lipid–AD pathway. Mediators were required to meet four criteria: (1) a significant causal association with AD; (2) a significant causal association with the lipid exposure; (3) a direct causal effect on AD independent of lipids; and (4) a mediating effect direction consistent with the total effect. In preliminary analyses, we estimated the total effect of lipids on AD (βC). Based on this, the mediation analysis proceeded in three steps: First, two-sample MR was performed to identify circulating metabolites with significant causal effects on AD and to estimate the causal effect of lipids on these metabolites (βA). Second, MVMR was conducted to estimate the direct causal effect of each metabolite on AD (βB) after adjusting for lipid exposure. Finally, we calculated the mediated effect (βA×βB), the direct effect (βC − βA×βB), and the proportion mediated [(βA×βB)/βC]. The 95% confidence intervals for the mediated effects and mediation proportions were estimated using the Delta method.
All analyses were conducted using R software version 4.5.0. The “TwoSampleMR” (version 0.6.16), “MVMR” (version 0.4.2) and “MendelianRandomization” (version 0.10.0) packages were used for MR analyses.
Results
Selection of instrumental variables
For the 179 lipid traits, 4506 SNPs were selected, with F-statistics ranging from 18.797 to 1969.067. From the 233 circulating metabolites, 13,121 SNPs were selected, with F-statistics ranging from 30.296 to 13,209.421. For AD treated as the exposure, 60 SNPs were extracted, with F-statistics ranging from 491.189 to 9769.809. All SNPs had F-statistics >10, indicating no bias arising from weak instrumental variables (Supplemental Tables 2–4).
Two-sample MR analysis of lipids on AD
To investigate the potential causal relationship between lipids and AD risk, we first performed two-sample MR analyses. Then, outliers were detected and removed using MR-PRESSO and Radial MR. After Bonferroni correction (p < 0.05/179 = 2.79 × 10−4), IVW estimates showed that only PI(18:1/18:1) (OR = 0.917; 95% CI = 0.884–0.951; p = 3.54 × 10−6) and TAG(52:3) (OR = 0.919; 95% CI = 0.889–0.949; p = 3.53 × 10−7) demonstrated significant associations with AD, both suggesting reduced AD risk (as shown in the Figure 2). The MR estimates were directionally consistent across all analytical methods, and no evidence of pleiotropy or heterogeneity was detected in sensitivity analyses (Supplemental Table 6A). Visualizations for Radial MR, leave-one-out analyses, and scatter plots are provided in the Supplemental Material.
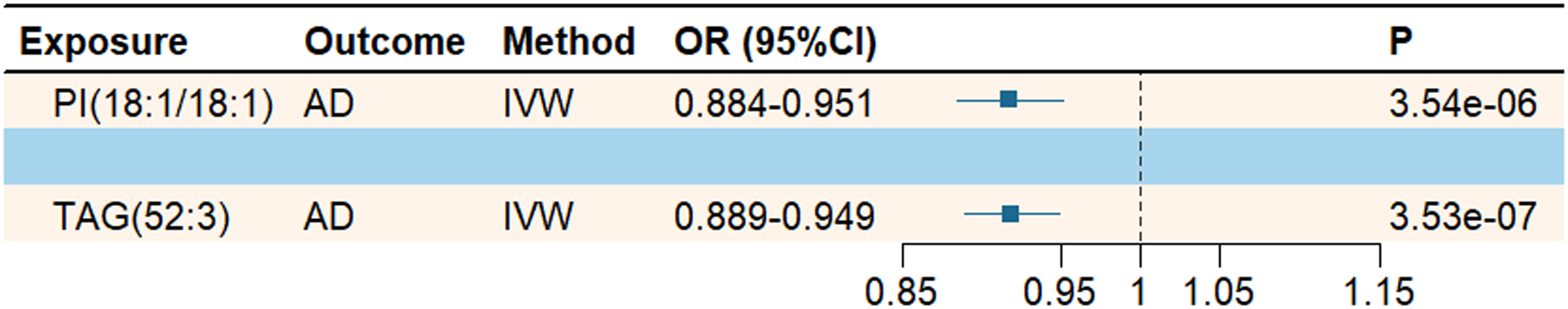
Mendelian randomization analysis of the causal relationship between lipids and Alzheimer's disease.
Reverse causality and genetic correlation analyses between lipids and AD
To eliminate potential reverse causation bias, we performed Steiger directionality tests and reverse MR analyses for PI(18:1/18:1) and TAG(52:3). The Steiger test indicated that the variance explained by the IVs for AD was markedly lower than that for the lipid traits, supporting a causal direction from lipids to AD (Supplemental Table 8A). Furthermore, in the reverse MR analysis, the IVW results showed no causal relationship between AD and either PI(18:1/18:1) or TAG(52:3) (p > 0.05) (Supplemental Table 7). Sensitivity analyses revealed no evidence of pleiotropy or heterogeneity (Supplemental Table 6B). LDSC analyses showed no significant genetic correlation between PI(18:1/18:1) (Rg = 0.050; SE = 0.138; p = 0.717), TAG(52:3) (Rg = 0.045; SE = 0.117; p = 0.704), and AD (Supplemental Table 8B). These findings suggest that the MR results are unlikely to be biased by shared genetic architecture.
Confounder analysis
To enhance the stability of the analytical results, we examined pleiotropic pathways related to AD to exclude the influence of confounding factors. In the MVMR analysis, we adjusted for three common confounders (alcohol consumption, physical activity, and sedentary behavior). The results showed that PI(18:1/18:1) and TAG(52:3) remained significantly associated with AD (p < 0.05), consistent with the causal associations identified in the primary MR analysis (as shown in the Table 1).
MVMR analysis of lipids on Alzheimer's disease.

Meta-analysis
To further validate the potential causal associations between PI(18:1/18:1), TAG(52:3), and AD, we repeated the MR analyses using AD datasets from independent sources. Subsequently, we conducted a meta-analysis to combine the IVW estimates obtained from the two analyses. As shown in the Figure 3, the meta-analysis results for PI(18:1/18:1) and TAG(52:3) both remained statistically significant (p < 0.05), with effect directions consistent with those in the primary MR analysis. These findings support the robustness and reliability of our MR results.
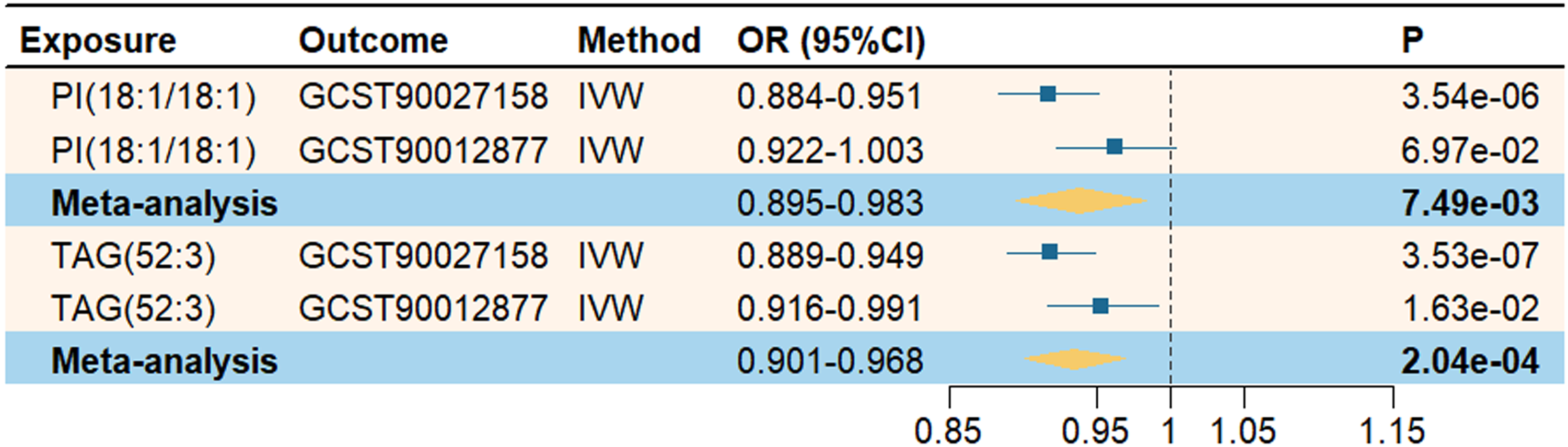
Meta-analysis of the causal relationship between lipids and Alzheimer's disease.
Mediation analysis of circulating metabolites
In exploring potential mediators, we initially selected 233 circulating metabolites to evaluate their effects on AD (Supplemental Tables 9 and 10). After sensitivity analyses and Bonferroni correction, IVW results showed that 29 metabolites were significantly associated with AD risk (p < 2.15 × 10−4 = 0.05/233), as illustrated in the Figure 4. Then, we examined the causal relationships between PI(18:1/18:1), TAG(52:3), and the AD-associated metabolites. The results revealed 17 causally associated metabolites when PI(18:1/18:1) was used as the exposure, and 8 when TAG(52:3) was the exposure (Supplemental Tables 11 and 12). Finally, we performed mediation MR to evaluate how PI(18:1/18:1) and TAG(52:3) influence AD via circulating metabolites, and calculated the mediated effects and proportions (Supplemental Table 13). The results indicated that PI(18:1/18:1) may reduce AD risk through increasing triglyceride levels in low-density lipoproteins (LDL; β = 0.176; 95% CI = 0.036–0.316), triglycerides in medium LDL (β = 0.228; 95% CI = 0.064–0.392), and triglycerides in small LDL (β = 0.181; 95% CI = 0.025–0.337). TAG(52:3) may reduce AD risk by decreasing the free cholesterol to total lipids ratio in medium high-density lipoproteins (HDL; β = 0.130; 95% CI = 0.015–0.244), as shown in Figure 5. All analyses passed the sensitivity checks, with no evidence of pleiotropy or heterogeneity detected.
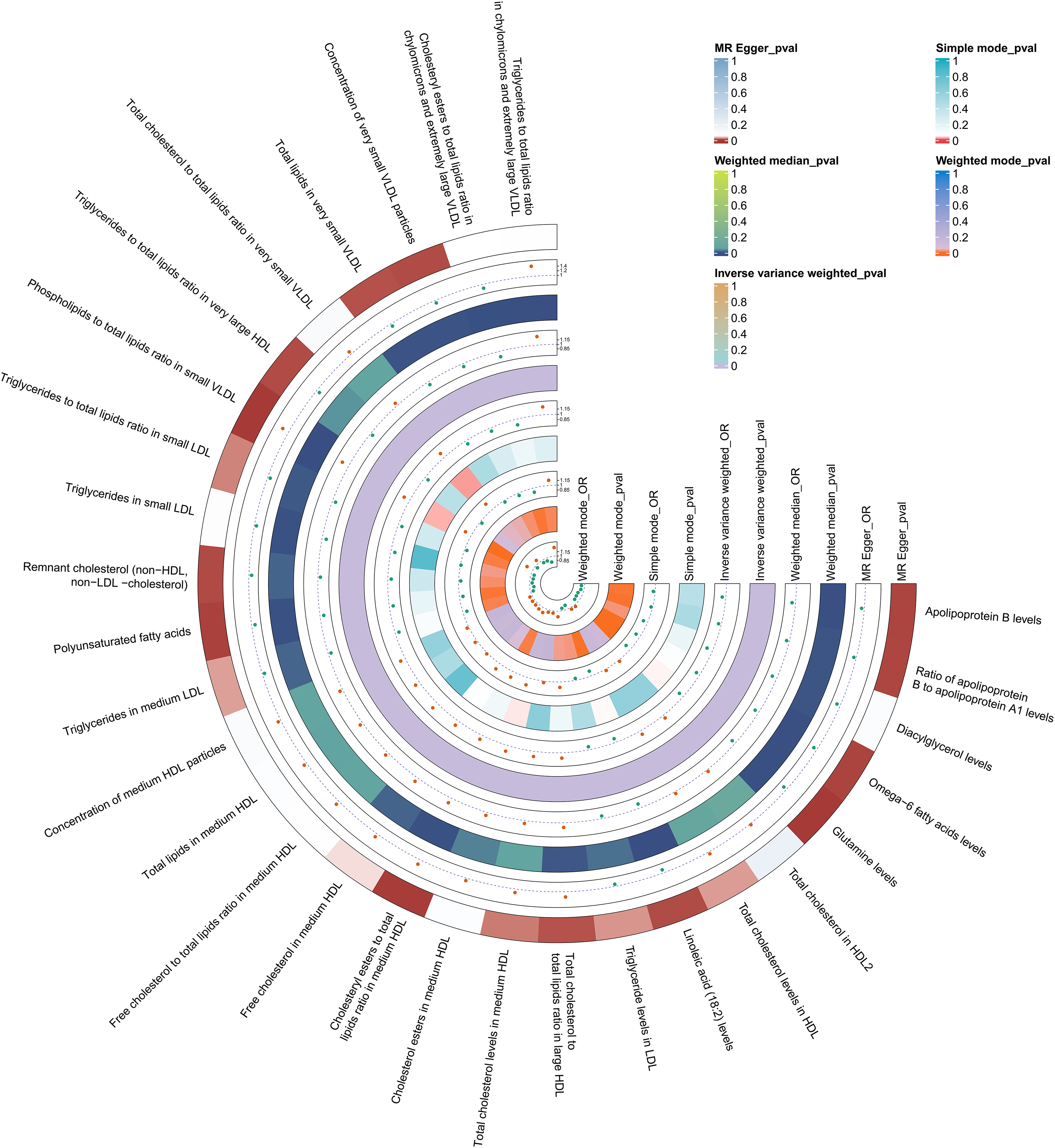
Mendelian randomization analysis of the causal relationship between circulating metabolites and Alzheimer's disease.
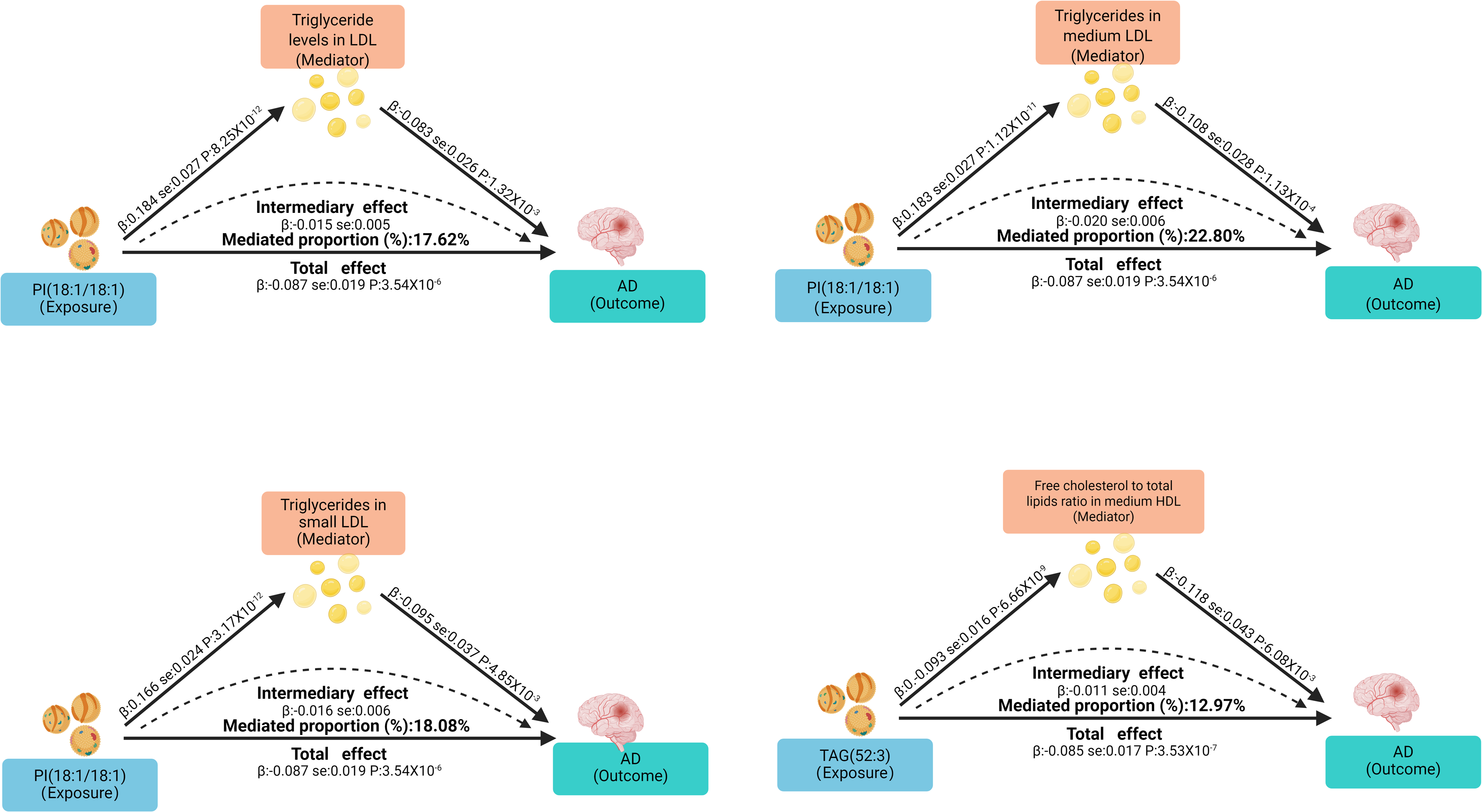
Mediating role of circulating metabolites in the effect of lipids on Alzheimer's disease.
Discussion
In this MR study, we investigated the causal effects of lipids on AD and the potential mediating roles of circulating metabolites. The results showed that higher levels of the lipids PI(18:1/18:1) and TAG(52:3) were associated with a reduced risk of AD. Further mediation analyses indicated that triglyceride levels in LDL, medium LDL, and small LDL are protective metabolites for AD, and PI(18:1/18:1) may reduce AD risk by increasing these metabolite levels, mediating 17.6%, 22.8%, and 18.1% of its protective effect, respectively. Additionally, the free cholesterol to total lipids ratio in medium HDL was identified as a risk-related metabolic indicator for AD, and TAG(52:3) may reduce AD risk by lowering this ratio, with a mediated proportion of 13%. These findings provide new insights into the role of lipid metabolism in AD pathogenesis and suggest that lipid-related pathways may serve as important molecular targets for future interventions and therapies.
Phospholipids are key components of cell membranes and myelin, and are primarily classified into glycerophospholipids and sphingolipids. Among them, phosphatidylinositol (PI) is one of the most abundant glycerophospholipids and plays a crucial regulatory role in multiple signal transduction pathways mediated by neurotransmitters and growth factors. 36 PI(18:1/18:1) is 1,2-dioleoyl-phosphatidylinositol, in which both the sn-1 and sn-2 positions are occupied by oleic acid (OA). Studies have shown that PI levels are significantly reduced in the hippocampus and cortex of patients with mild cognitive impairment (MCI) and AD. 12 Moreover, PI downregulation accelerates Aβ aggregation and impairs neurotransmitter release, thereby promoting AD pathological progression. 37 The OA component of PI(18:1/18:1) also exerts beneficial effects in AD. Amtul et al. 38 reported in a transgenic AD mouse model that OA markedly reduces Aβ42 levels and decreases amyloid plaque formation; subsequent studies further demonstrated that elevated plasma OA levels are associated with reduced risk of AD and MCI, and are closely linked to slower progression from MCI to AD. Taken together, these findings suggest that PI(18:1/18:1) may serve as a potential biomarker for early detection of AD.
Triacylglycerols (TAGs) are the most abundant glycerolipids in plasma and constitute a major proportion of total lipids. The relationship between TAG levels and AD risk remains inconclusive across studies. Some studies have reported that elevated TAG levels in midlife are associated with later-life cognitive decline. 39 Conversely, Zhou et al. 40 observed in a community-based cohort aged ≥65 years that higher plasma TAG levels were associated with a lower risk of dementia and slower cognitive decline. TAG(52:3) is a moderately unsaturated triglyceride, and its fatty acid composition confers distinct biological functions. Tajima et al. 41 reported that in APP/tau double-transgenic AD mice, TAG(60:12) levels were elevated at 10 months, whereas TAG(52:3) levels were significantly reduced at 15 months, compared with wild-type controls. This finding suggests that the impact of TAG metabolism on AD may be jointly regulated by age stage and molecular subtype. Therefore, different TAG subtypes may exert opposing effects at early versus late stages of the disease, indicating the need for future studies to investigate their stage-specific roles and underlying pathophysiological significance in AD.
LDL contain a core composed of cholesteryl esters (CE) and TAGs, surrounded by a surface monolayer of phospholipids, free cholesterol, and apolipoprotein B-100. 42 CE transfer protein mediates the exchange of CE in HDL with TAG in LDL, thereby increasing LDL cholesterol (LDL-C) levels and reducing LDL-TAG content. 43 Previous studies have reported that such metabolic alterations may be associated with cognitive decline and an increased risk of dementia.44,45 Although direct evidence supporting a protective effect of triglyceride levels in LDL on AD is still lacking, existing findings suggest that higher LDL-TAG levels may indirectly reduce AD risk by suppressing LDL-C accumulation. Moreover, PI(18:1/18:1) can inhibit p38 mitogen-activated protein kinase activation and endoplasmic reticulum stress, maintain membrane lipid unsaturation and cell morphology, and thereby promote lipid homeostasis. 46 Taken together, the protective effect of PI(18:1/18:1) on AD may be partially mediated by triglyceride levels in LDL, a hypothesis consistent with our MR findings.
HDL is a complex lipoprotein particle composed of various lipids (CE, TAG, and phospholipids) and proteins (apolipoproteins and enzymes). The functions of HDL vary depending on its composition and quality. 47 Among its functions, cholesterol efflux capacity is primary and plays a key role in preventing cardiovascular disease, stroke, and dementia.48,49 However, an increased proportion of free cholesterol in HDL typically indicates insufficient cholesterol esterification, which impairs its cholesterol efflux capacity. 50 Significant reductions in cholesterol efflux capacity have been observed in patients with MCI and AD. 51 Additionally, animal studies have further demonstrated that impaired HDL cholesterol efflux promotes Aβ plaque deposition in the brain and leads to cognitive decline. 52 Therefore, an elevated free cholesterol to total lipids ratio in medium HDL may reflect impaired HDL function and thereby increase AD risk. Moreover, Liu et al. 53 reported that elevated plasma TAG levels can alter the composition and structure of HDL. Taken together, TAG(52:3) may exert protective effects against AD by lowering the free cholesterol to total lipids ratio in medium HDL and thereby improving HDL cholesterol efflux function. This is consistent with our MR results.
This study has several notable strengths. We leveraged GWAS datasets from multiple consortia to investigate the causal relationship between lipids and AD, thereby minimizing potential sample overlap. The MR framework was implemented under rigorous statistical principles, incorporating multiple sensitivity analyses to ensure the robustness of the findings. In addition, Steiger directionality tests and reverse MR were applied to rule out reverse causation. LDSC analysis helped avoid bias arising from genetic correlation. MVMR analysis controlled for common confounding factors. Replication analyses and meta-analytic validation further strengthened the study's findings, and a two-step MR framework was introduced to explore circulating metabolites as mediators. Nevertheless, this study also has several limitations. First, the data were primarily derived from European populations, limiting the generalizability of the findings to other ethnic groups. Second, the lack of individual-level data prevented stratified analyses by age or sex. Third, inherent limitations of MR remain; although confounders were adjusted for using MVMR and sensitivity analyses showed no major heterogeneity or horizontal pleiotropy, undetected pleiotropy cannot be completely excluded. Finally, the causal inferences drawn from MR analyses require further validation and supplementation through animal studies and randomized controlled trials.
Conclusion
The MR findings of this study revealed causal effects of the lipids PI(18:1/18:1) and TAG(52:3) on AD. Further mediation analyses indicated that triglyceride levels in LDL, medium LDL, and small LDL, as well as the free cholesterol to total lipids ratio in medium HDL may exert potential mediating effects in this process. These findings provide new insights into the role of lipid metabolism in AD pathogenesis and suggest that lipid-related pathways may serve as potential targets for prevention and intervention.
Supplemental Material
sj-docx-1-alr-10.1177_25424823261456655 - Supplemental material for Circulating metabolites mediate the effects of lipids on Alzheimer's disease: A Mendelian randomization and mediation analysis
Supplemental material, sj-docx-1-alr-10.1177_25424823261456655 for Circulating metabolites mediate the effects of lipids on Alzheimer's disease: A Mendelian randomization and mediation analysis by Xinyu Yang, Chang Chai, Wenjing Li and Yanjie Li in Journal of Alzheimer's Disease Reports
Supplemental Material
sj-xlsx-2-alr-10.1177_25424823261456655 - Supplemental material for Circulating metabolites mediate the effects of lipids on Alzheimer's disease: A Mendelian randomization and mediation analysis
Supplemental material, sj-xlsx-2-alr-10.1177_25424823261456655 for Circulating metabolites mediate the effects of lipids on Alzheimer's disease: A Mendelian randomization and mediation analysis by Xinyu Yang, Chang Chai, Wenjing Li and Yanjie Li in Journal of Alzheimer's Disease Reports
Footnotes
Acknowledgements
We gratefully acknowledge the investigators, participants, and data platforms that made the GWAS datasets used in this study publicly available.
Ethical considerations
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Administration of Traditional Chinese Medicine Science and Technology Project “Zhang Zhongjing Inheritance and Innovation Special Project” (GZY-KJS-2022-043-3) and Traditional Chinese Medicine Discipline Project of Henan Province's “Double First Class” Creation Project (HSRP-DFCTCM-2023-3-11).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets analyzed during the current study are publicly available from the original genome-wide association studies, public repositories, consortia, and published sources cited in the manuscript. Details of the data sources and access information are provided in the Methods section and reference list.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.