
Abstract
Background
Parkinson's disease (PD) is marked by the progressive loss of dopaminergic neurons in the substantia nigra and the accumulation of α-synuclein (α-syn) into amyloid fibrils and Lewy bodies. These aggregates impair synaptic function, disrupt proteostasis, compromise mitochondrial health, and drive neuroinflammation and oxidative stress. Clinical observations during the COVID-19 pandemic revealed accelerated motor deterioration and higher mortality among PD patients following SARS-CoV-2 infection, pointing to potential overlapping mechanisms with Alzheimer's disease, including protein misfolding, chronic inflammation, and oxidative damage.
Objective
This study aims to investigate how the SARS-CoV-2 N-protein may accelerate α-syn aggregation using computational approaches.
Methods
We employed a multimodal, label-free computational pipeline inspired by TopoBind to examine the topological maturation of patient-derived α-syn fibrils across preclinical (PDB 7V47, 8H03), mid-stage (7XO0, 7XO1), and late-stage (8H04, 7V48) PD phases. Per-residue features were extracted using 640-dimensional evolutionary scale modeling (ESM)-2 embeddings, solvent-accessible surface area, and persistent homology based on Vietoris–Rips filtration of Cα coordinates (r = 0–20 Å, persistence threshold > 0.8 Å). Predictions were validated using PyRosetta InterfaceAnalyzerMover, PRODIGY (ΔG and Kd), and normal mode analysis.
Results
The pipeline successfully identified key protein-protein interface residues on α-syn fibrils, achieving 74–92% overlap with PyRosetta results. Conserved interaction hotspots were located primarily in the non-amyloid component region, protofilament interfaces, and hydrophobic grooves.
Conclusions
Our findings demonstrate that fibril maturation involves progressive expansion of binding interfaces, offering a plausible topological mechanism by which SARS-CoV-2 N-protein can promote pathogenic α-syn conformers and contribute to long-term neurological complications following infection.
Keywords
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder marked by the selective loss of dopaminergic neurons in the substantia nigra pars compacta, resulting in cardinal motor symptoms such as bradykinesia, rigidity, resting tremor, and postural instability, along with various non-motor features. At the molecular level, PD is driven primarily by the misfolding and aggregation of α-synuclein (α-syn), a presynaptic protein that forms insoluble amyloid fibrils and Lewy bodies in affected brain regions. These aggregates disrupt cellular homeostasis, promote neuroinflammation, impair mitochondrial function, induce oxidative stress, and compromise protein clearance pathways, ultimately leading to neuronal death.1,2Similar protein misfolding and aggregation mechanisms, together with shared pathways of neuroinflammation, oxidative stress, mitochondrial dysfunction, and impaired proteostasis, are implicated in other major neurodegenerative diseases such as Alzheimer's disease, where amyloid-β and tau pathologies contribute to neuronal loss and cognitive decline. Insights into how external triggers like viral infections may accelerate these common processes of neurodegeneration thus hold relevance for advancing understanding and progress in Alzheimer's disease as well.
The term parkinsonism refers to a clinical syndrome characterized by bradykinesia plus at least one of rigidity, resting tremor, or postural instability. Although idiopathic PD is the most common cause, parkinsonism can also arise from other neurodegenerative, vascular, drug-induced, or infectious etiologies. In this study, “PD” denotes idiopathic Parkinson's disease, whereas “parkinsonism” refers to the broader clinical presentation, including cases potentially triggered or accelerated by external factors such as viral infections.
The etiology of PD is multifactorial, involving genetic predispositions (e.g., mutations in SNCA, LRRK2, and PARKIN), environmental exposures, and aging. Recent evidence increasingly highlights the role of infectious agents in modulating disease onset and progression.1–2 The emergence of SARS-CoV-2, the causative agent of COVID-19, has introduced a new dimension to PD research. Beyond its primary respiratory effects, SARS-CoV-2 infection is associated with a broad spectrum of neurological complications, including encephalitis, Guillain-Barré syndrome, and cerebrovascular events. 3 Clinical observations during the pandemic showed that individuals with preexisting PD often experienced accelerated motor decline and increased mortality. Cohort studies and meta-analyses reported an elevated risk of neurodegenerative disorders, including PD (hazard ratio 1.44, 95% CI: 1.06–1.95).4–5 Case reports and longitudinal data have also documented new-onset Parkinsonism following SARS-CoV-2 infection6–8
Mechanistically, the SARS-CoV-2 nucleocapsid N-terminal domain (N-NTD) binds α-syn directly, accelerating amyloid fibril formation and enhancing seeding efficiency in vitro.9–11 Preclinical models further demonstrate that SARS-CoV-2 exposure exacerbates α-syn aggregation, dopaminergic toxicity, and neuroinflammation.12–13 These findings suggest that SARS-CoV-2 may act as an environmental trigger that promotes or accelerates α-syn pathology, possibly by exploiting maturation-dependent structural features of fibrils. 14
To investigate this interaction, patient-derived α-syn fibril polymorphs were categorized into three stages based on clinical and pathological progression: pre-PD: preclinical phase (asymptomatic or before motor symptom onset; e.g., PDB 7V47, 8H03); mid-PD: middle-to-late clinical stages with moderate to advanced motor symptoms (e.g., PDB 7XO0, 7XO1); and late-PD: advanced or terminal stage of PD, often with severe motor deficits and dementia (e.g., PDB 8H04, 7V48).15,16 These stages reflect increasing conformational maturation and pathological severity observed in cryo-EM studies.
We employed advanced computational approaches, including persistent homology, multifractal analysis, and a label-free multimodal interface prediction pipeline (TopoBind), to characterize the topological evolution of α-syn fibrils across these stages and their interactions with the SARS-CoV-2 N-protein. The overall aim is to elucidate potential molecular mechanisms by which SARS-CoV-2 infection may accelerate α-syn pathology and PD progression, while providing a computational framework for identifying stage-specific therapeutic targets. 17
Methods
This section details the TopoBind pipeline, a label-free, structure-based computational framework for predicting protein-protein interaction (PPI) interface residues. TopoBind integrates topological data analysis18,19 (persistent homology), surface exposure metrics (SASA), 20 sequence-derived contextual embeddings (from ESM-2), 21 and graph-based learning (GraphSAGE) to identify binding interfaces without requiring supervised training on annotated interface labels, as shown in Figure 1. The method is particularly suited for exploratory analyses of novel or understudied complexes, such as those between pathological α-syn fibril polymorphs and the SARS-CoV-2 (N-NTD).

Methodology.
Protein structures and preparation
α-syn fibril structures were obtained from the Protein Data Bank: pre-PD polymorphs 7V47 and 8H03, mid-PD (7XO0 and 7XO1), and late-PD (7V48 and 8H04). This is a computational study that used publicly available cryo-EM structures of α-syn fibrils from the Protein Data Bank. No new human subjects or biological samples were collected for the present work. These structures originate from cerebrospinal fluid (CSF) of one preclinical PD patient and one late-stage postmortem PD patient in a prior study conducted at Huashan Hospital, Fudan University, Shanghai, China. The SARS-CoV-2(N-NTD) (residues 1–180) was obtained from PDB entry 6M3M. All structures were processed using Biopython: non-protein atoms were removed, alternate conformations were resolved by selecting the highest-occupancy positions, and residues were renumbered sequentially for consistency across chains.
Active residue prediction and docking
Potential active residues on α-syn fibrils were identified using ISPRED4 22 (Interaction Site PREDictor version 4) with a probability threshold ≥ 0.5 and relative SASA > 20%. Protein–protein docking was performed on the HADDOCK2.4 (High Ambiguity Driven protein-protein DOCKing) web server. 23 Ambiguous interaction restraints were derived from ISPRED4 active residues on the fibril, and the viral protein was docked in fully ambiguous mode. The standard protocol was followed: 1000 rigid-body docking models, 200 semi-flexible refinement models, and 200 explicit-solvent refined models. The top-ranked cluster (with the lowest HADDOCK score) was selected for each polymorph.
The scoring is performed according to the weighted sum (HADDOCK score) of the following terms:
Evdw: van der Waals intermolecular energy Eelec: electrostatic intermolecular energy Eair: distance restraints energy (only unambiguous and AIR (ambig) restraints) BSA: buried surface area
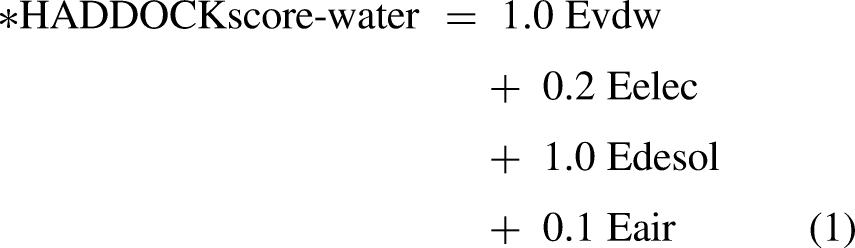
Multimodal feature extraction
In the TopoBind pipeline, feature extraction is a crucial step that converts raw structural information from the input protein complex (PDB file) into comprehensive per-residue feature vectors. These features integrate sequence context, surface properties, and topological characteristics for each residue in both chains (A and B). The resulting multimodal descriptors are then used as node features to construct residue-level graphs for subsequent processing by the graph neural network (GNN).
Sequence extraction and embeddings: The primary amino acid sequence serves as the basis for creating advanced embeddings (via ESM-2). It encodes the biochemical identity of each residue. The sequence is assembled by concatenating peptides from the chain, ignoring hetero-residues or gaps. To represent protein sequences in our pipeline, we use per-residue embeddings derived from ESM-2, a robust transformer-based protein language model developed by Meta AI's Fundamental AI Research team.21
19
ESM-2 is pre-trained on extensive protein sequence databases (mainly UniRef clusters) using a masked language modelling objective. During training, random amino acids are masked, and the model learns to reconstruct them using surrounding context. The training loss is cross-entropy over the masked positions.
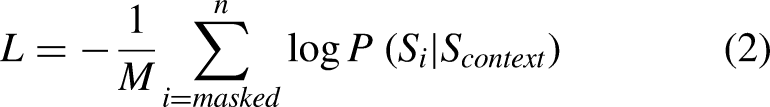
This process enables the model to capture rich evolutionary, structural, and functional patterns encoded in protein sequences. The core architecture relies on the standard scaled dot-product attention mechanism:

Persistent homology: Persistent homology is a powerful method in topological data analysis that examines the shape of data, such as collections of points in space, by detecting and measuring topological structures (connected pieces, rings, cavities, etc.) across different distance scales. We used persistent homology to analyze the backbone geometry of each protein chain. It processes the 3D positions of the alpha-carbon (Cα) atoms a sparse but representative set of points that traces the protein's overall fold. The goal is to identify residues near stable topological features (such as persistent loops or enclosed regions) that are often associated with binding sites or functionally important regions on protein–protein interfaces. The computation uses the Ripser library, which implements an efficient algorithm for Vietoris–Rips persistent homology on point clouds. Below is a step-by-step breakdown of the mathematical concepts. Input Point Cloud: Point Cloud(X) is a discrete set of points representing the backbone structure of a protein chain. Specifically, this is the collection of three-dimensional coordinates for the alpha-carbon (Cα) atoms, which serve as a simplified proxy for the protein's overall fold and geometry. let In proteins, Cα atoms are spaced approximately 3.8 Å apart along the backbone, forming a quasi-linear but folded structure. The choice of Cα over all atoms reduces dimensionality and focuses on the global topology, making it suitable for persistent homology, where local atomic fluctuations (e.g., from thermal motion or PDB resolution limits of ∼1–3 Å) could otherwise generate spurious, short-lived features. Vietoris–Rips filtration: To analyze the multi-scale topology of the point cloud X, constructs a filtered simplicial complex. This VR complex at a given scale r is defined as the abstract simplicial complex where a k-simplex [x{i0}, …, x{ik} ] is included if the diameter of the subset is at most 2r, This creates a nested sequence of complexes, known as a filtration: Homology computation: Persistent homology groups up to dimension 1, focusing on 0-dimensional (H_0: connected components) and 1-dimensional (H_1: loops or cycles) features, which are most informative for protein backbones. For a simplicial complex K, homology in dimension k quantifies k-dimensional “holes”. The key algebraic objects are defined as follows: The The A cycle is a chain z ∈ Ck(K) such that ∂k(z) = 0 — that is, a closed loop or surface with no boundary.




A boundary is a chain b ∈ Ck(K) that is the image of the boundary operator from a higher dimension, b = ∂k+1(c) for some c ∈ Ck+1(K) — meaning it is the “edge” of a filled higher-dimensional simplex
The
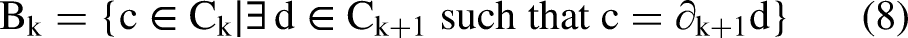


The matrix reduction (using a persistent co-homology variant for speed) on the filtered boundary matrix. Simplices are ordered by their birth time in the filtration. For H0 (dimension 0): starts with β0 = N isolated points, then decreases as edges appear and merge components. For H1 (dimension 1): β1 increases when independent cycles (loops) are born, then decreases when those cycles are filled by higher simplices (e.g., triangles closing a loop).
Persistence diagram: In the persistence diagram, each topological feature is represented by a pair (b, d) where b is the birth time (filtration value where the class emerges) and d is the death time (where it vanishes) A homology class [γ] ∈ Hk(VR(b)) is The class In the persistent homology analysis of protein Cα backbones, typical filtration scales reflect the structural hierarchy of the polypeptide chain. For dimension 0 (H0, connected components), births occur at small values (∼0–3 Å), corresponding to individual residues appearing as isolated points. Deaths follow shortly afterward (∼3–10 Å), as edges form and merge these points into clusters representing secondary structure elements such as helices and sheets. For dimension 1 (H1, loops and cycles), births generally appear at intermediate scales (∼5–15 Å), marking the emergence of closed rings or loops in the backbone trace—often associated with turns, hairpins, or pocket-like features relevant to binding sites. Deaths occur at larger filtration values, when sufficient higher-dimensional simplices (e.g., triangles or tetrahedra) fill in these loops, causing them to become boundaries of higher chains. • Persistence filtering: To filter out noise, we applied a persistence threshold, retaining only topological features with persistence π = d − b > 0.8 Å. This threshold discards short-lived bars likely caused by coordinate noise, sampling artifacts, or PDB resolution limits (backbone RMSD typically 0.5–1 Å), while retaining robust topological signals. The 0.8 Å cutoff is an empirical choice suited to protein Cα data: features below it is usually transient and biologically insignificant, whereas longer persistence (e.g., π ≈ 5–10 Å for stable H1 loops in beta-hairpins) often corresponds to meaningful structures like binding pockets or functional cavities. Mathematically, the persistence diagram Dgm_k = {(bi, di)} is restricted to the subset where πi > 0.8, yielding a concise list of triples (dim, birth, death). This removes many short H0 bars (transient clusters) and preserves durable H1 features (persistent loops), improving the biological interpretability of the extracted topological residues. • Mapping persistence features to residues: We mapped each persistent topological feature (dim, b, d) to nearby residues by first calculating the midpoint scale center_dist = (b + d)/2, which approximates the radius at which the feature is most prominent. For every residue i, it checks whether the distance from its Cα position xᵢ to the chain centroid μ = (1/N) ∑ xj (i.e., coords.mean(0)) satisfies ‖xᵢ − μ‖2 < center_dist + 5.0 Å; if so, residue i is added to topo_set. This creates a spherical region around the chain's center-of-mass, expanded by a 5 Å buffer to accommodate backbone spacing (∼3.8 Å between Cα atoms), side-chain reach (∼2–5 Å), and minor geometric offsets, ensuring inclusion of residues bordering the feature (e.g., those lining a persistent loop or pocket). These topological residues are then used as binary node features in the residue graphs and receive a + 0.12-score bonus in the final predictions, thereby providing a geometric prior that biases the GNN toward interface-relevant motifs.
for small ɛ > 0. This means the feature (e.g., a new loop) emerges when simplices are added at scale ≈ b/2 (since filtration value = 2r in Vietoris–Rips distance terms).
sends [γ] → 0, while

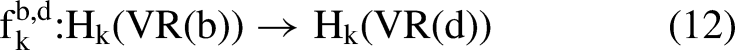

SASA calculation: SASA was calculated using FreeSASA. For each specified chain in a PDB file, the structure was loaded and SASA computed with default parameters (Lee-Richards algorithm, 1.4 Å probe radius). Total SASA values per residue were extracted and stored in a dictionary with Biopython-compatible residue IDs. Results were cached as pickled files by PDB and chain to enable fast reuse.
Residue graph construction
For each protein chain, per-residue features from three complementary sources ESM-2 sequence embeddings, solvent accessible surface area values, and topological flags from persistent homology were concatenated to produce a high-dimensional multimodal descriptor per amino acid (total dimensionality ≈ 1500–1800). A single linear projection layer mapped these concatenated vectors into a unified 512-dimensional embedding space, ensuring compatibility for graph-based learning. Residue-level graphs were then constructed, with nodes corresponding to individual amino acids and node features set to the projected 512-dimensional vectors. Edges included two types: (i) spatial edges connecting each residue to its k = 15 nearest neighbors based on Cα–Cα Euclidean distance (with a hard cutoff of 12 Å to limit long-range noise), and (ii) sequential edges linking each residue i to residues i ± 1 and i ± 2 along the polypeptide chain. Edge attributes encoded the raw Euclidean distance for spatial neighbours and the absolute sequence separation (|i − j|) for sequential connections, providing both geometric and sequential context to the message-passing layers.
Interface residue prediction
Interface residue prediction was performed using a lightweight GraphSAGE model. The model consisted of three layers employing mean aggregation, each with a hidden dimension of 256, ReLU activations, and dropout of 0.3 for regularization. The output of the final layer was fed into a single linear head that produced a scalar logit per residue. During inference, these logits were passed through a sigmoid function to yield interface probability scores in [0, 1].
Unlike many supervised approaches, the TopoBind pipeline did not rely on pre-trained weights or external contrastive pretraining on generic protein interface datasets. Instead, the model was trained from scratch (random initialization) on the target complex itself in a self-supervised manner: binary ground-truth labels were derived directly from the 5 Å inter-atomic contact criterion between the two chains in the provided PDB file. This per-complex training loop (80 epochs, Adam optimizer lr = 0.001, BCEWithLogitsLoss with pos_weight=5.0 to handle imbalance) allowed the model to learn a highly accurate, structure-specific classifier tailored to the exact geometry and features of the input complex.
Validation and downstream analyses
PyRosetta4 24 was used to orthogonally validate the interface residues predicted by TopoBind by evaluating per-residue energetic contributions in the bound α-syn fibril–SARS-CoV-2 N-NTD complexes. This physics-based approach identifies residues that strongly stabilize the complex within the Rosetta full-atom energy landscape, providing independent energetic confirmation of the interface's relevance. The analysis was performed on the docked complexes 7v47_6m3m.pdb, 7v48_6m3m.pdb, 8h04_6m3m.pdb, and 8h03_6m3m.pdb (with chain A corresponding to the fibril protomer and chain B to the N-NTD).
For every residue in the identified interface set, the chain identifier and three-letter amino acid code were recorded, and the total per-residue energy was extracted using pose.energies().residue_total_energy(res). This comprehensive energy term encompasses all intra-residue, inter-residue, solvation, hydrogen bonding, van der Waals, electrostatic, and environment-dependent contributions in the bound state. Residues with total energy ≤ −0.5 REU were classified as energetically favourable contributors and sorted by the most negative (most stabilizing) values.
Summary statistics reported the total number of contributors and the top 5 most favourable residues, as shown in Table 6. Across fibril polymorphs, this procedure typically identified 10–40 contributing residues per complex, with the strongest contributors exhibiting total energies ranging from −2.0 to −5.0 REU or lower, consistent with the energetic profile of key interface hotspots involved in critical packing, hydrogen bonding, or hydrophobic interactions.
Binding affinity prediction and energetic cross-validation
Binding affinities of the docked α-syn fibril–SARS-CoV-2 N-NTD complexes were estimated using PRODIGY,25–26 an empirical prediction tool that calculates the change in binding free energy (ΔG) and the corresponding equilibrium dissociation constant (Kd) at 25 °C using equations 14 and 15. The method relies on a linear regression model trained on experimentally determined affinities and incorporates the following structural descriptors extracted from the interface: the number and type of intermolecular contacts (including hydrogen bonds, salt bridges, and hydrophobic interactions), desolvation penalties, electrostatic contributions, and the non-interacting surface area. PRODIGY was applied directly to the docked PDB files without additional relaxation or minimization steps.
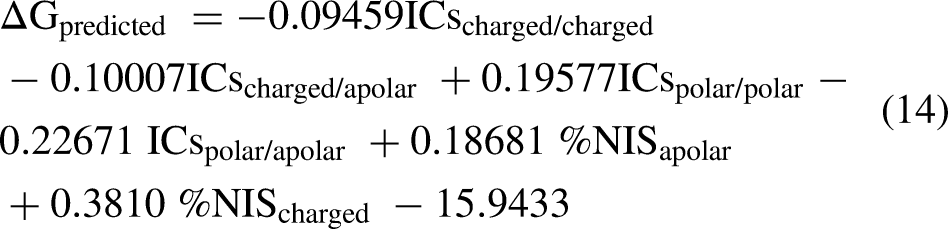
ICsxxx/yyy is the number of Interfacial Contacts found at the interface between Interactor1 and Interactor2 classified according to the polar/apolar/charged nature of the interacting residues (i.e., ICscharged/apolar is the number of ICs between charged and apolar residues) as shown in the equation 14. Two residues are defined in contact if any of their heavy atom is within a distance of 5.5 Å.
Based on the predicted binding affinity (ΔG) according to Equation 14, the dissociation constant (Kd) is calculated via the following equation 15 shown below:

To cross-validate the PRODIGY-predicted affinities, global and per-residue energetic metrics were computed independently using PyRosetta4. Each docked complex was loaded and scored using the full-atom energy function. Interface analysis was then performed for the chain pairing A_B (A = fibril chain, B = N-NTD chain), with options enabled for separate side-chain packing, input-structure packing, and packing-statistics computation. This analysis identified the set of interface residues and provided the global interface binding energy (dG separated) in Rosetta Energy Units (REU), together with the change in solvent-accessible surface area (ΔSASA) at the interface and the packstat score for structural quality assessment.
For local energetic assessment, the total per-residue energy was extracted for all residues within the interface set. Residues exhibiting total energy ≤ −0.5 REU were classified as favorable contributors to interface stability. These residues were sorted in descending order of stabilization (most negative values first). The procedure thus combines a fast, contact-based empirical affinity prediction (PRODIGY) with physics-based Rosetta interface energetics (global dG separated + per-residue total energies) to evaluate whether the predicted binding strength and energetic favorability differ systematically between PD-patient-derived and healthy-brain-derived fibril conformers.
Normal mode analysis
To elucidate intrinsic and binding-induced dynamics in the docked complexes, anisotropic network model (ANM) was applied using a custom implementation based on a coarse-grained elastic network representation using ProDy.
27
Cα atoms from both chains were selected as nodes, and pairs within a 15 Å cutoff were connected by harmonic springs with uniform strength (γ = 1.0). The total potential energy V for the network is calculated by using equation 16.
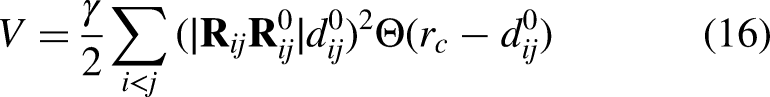
The Hessian matrix was constructed using equation 17 to encode the second derivatives of the harmonic potential, with off-diagonal 3 × 3 blocks representing negative spring forces between connected nodes and diagonal blocks the positive sum of attached springs.The 3N × 3N Hessian H (N = number of Cα atoms across both chains) has block form:
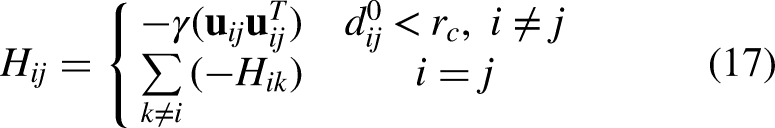
Eigen decomposition of the Hessian yielded normal modes using equation 18, from which the first 20 non-trivial modes (excluding six zero-frequency rigid-body modes) were used to compute root-mean-square fluctuations (RMSF) per residue and cross-correlation matrices between residue pairs, as shown in equations 19, 20, and 21.
Eigen decomposition:

Root-Mean-Square Fluctuation (RMSF): Per-residue fluctuation for Cα atom i:
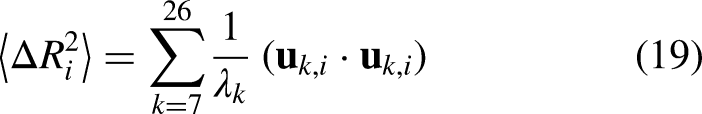
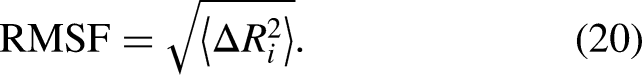
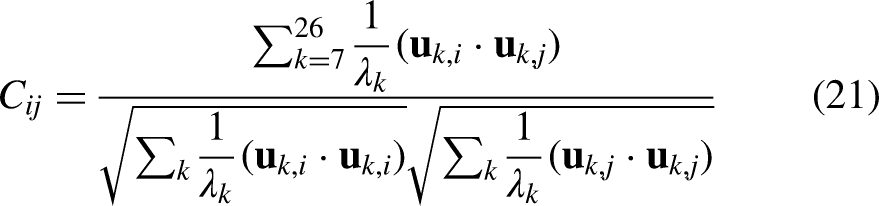
C_ij ranges from −1 (anti-correlated) to +1 (correlated).
RMSF values highlighted enhanced mobility in the N-terminal region (residues 1–50) of PD-patient-derived polymorphs (8H04, 7V48, 8H05), while cross-correlation matrices revealed strong inter-chain coupling (correlated and anti-correlated motions across the α-syn chain A and N-NTD chain B boundary) exclusively in pathogenic structures, absent in healthy polymorphs (7V47, 8H03).
Results
The α-syn fibril polymorphs representing Pre-PD (7V47, 8H03), mid-PD (7xo0 and 7xo1), and late-PD patient brain tissue (7V48, 8H04) were docked against the SARS-CoV-2 NTD (residues 1–180, PDB 6M3M) using HADDOCK2.4 with ambiguous interaction restraints derived from ISPRED4-predicted active residues on the fibril surface. The top-ranked cluster from each docking run was selected as the representative bound complex, as shown in Figure 2. The HADDOCK2.4 docking results show that SARS-CoV-2 NTD binds favorably to several α-syn fibril polymorphs. The healthy fibril complex 8H03_6m3m, as shown in Figure 2(a), ranks highest with a HADDOCK score of −126.3 ± 2.2 kcal/mol, low RMSD (5.4 ± 0.8 Å), strong electrostatics (−219.0 kcal/mol), and a large buried surface area (∼3010 Å2), as shown in Table 1. PD-associated complexes 8H04_6m3m and 7V48_6m3m, as shown in Figure 2(d) and 2(e), perform comparably or better in electrostatics (up to −252.8 kcal/mol) and interface size (>3000 Å2), suggesting a potential preference for pathological polymorphs. The healthy 7V47_6m3m is slightly weaker but still viable, as shown in Figure 2(b). In contrast, the PD CSF-seeded 7X01_6m3m, as shown in Figure 2(c), offers the largest interface (3533 Å2) and the best van der Waals (−115.9 kcal/mol) but is unreliable due to a poor overall score (−49.2 kcal/mol) and an extremely high restraint violation energy (1103 kcal/mol), indicating severe incompatibility with the input restraints. Overall, electrostatics dominate favorable binding in reliable models, with large interfaces supporting direct NTD-fibril interactions. These findings align with experimental evidence that the SARS-CoV-2 N protein promotes α-syn fibrillization, particularly in PD-relevant contexts.

(a-e) Docked complex of a-syn fibrils (Pre-PDd, Mid-PD, and Late-PD) and SARS-CoV2- N-protein (6m3m).
HADDOCK2.4 docking statistics for the top-ranked clusters of α-synuclein fibril–SARS-CoV-2 nucleocapsid NTD complexes.

Table 2 reports maximum persistent Betti numbers (max β0, max β1, max β2) from persistent homology analysis of patient-derived α-syn fibril structures across PD stages, based on cryo-EM-derived conformers for pre-PD (8H03 and 7V47), mid-PD (7XO0 and 7xo1), and late-PD (8H04 and 7V48), as shown in Figure 3. Max β0 increases modestly from 186–187 in pre-PD to 188 in mid-PD and 188–189 in late-PD, indicating progressive compaction and unification of the fibril topology as disease advances. Max β1 shows the strongest trend, rising from lower, more variable values of 63–81 in pre-PD to 82 in mid-PD and peaking at 81–85 in late-PD, reflecting enhanced persistence and abundance of 1D loops and cycles generated by maturing β-sheet ladders and a twisted cross-β architecture—signaling rigidification and stabilization of the amyloid core in later stages. Max β2 remains low (2 in pre-PD, increasing slightly to 3–4 in mid- and late-PD), consistent with dense fibril cores that enclose few voids, though the minor rise suggests the emergence of small peripheral pockets in mature forms. These topological features reveal a clear pattern of gradual structural maturation in α-syn fibrils during PD progression, with the dominant increase in max β1 highlighting strengthened β-sheet integrity and global shape complexity. These persistent Betti numbers are integrated into a multimodal machine learning pipeline—alongside SASA and ESM-2 embeddings—to predict interface residues in PPI, such as between α-syn and the SARS-CoV-2 N-protein, where they capture robust, multiscale shape invariants (especially loop-rich patterns at binding hotspots) to improve identification of contact residues in flexible amyloid systems.
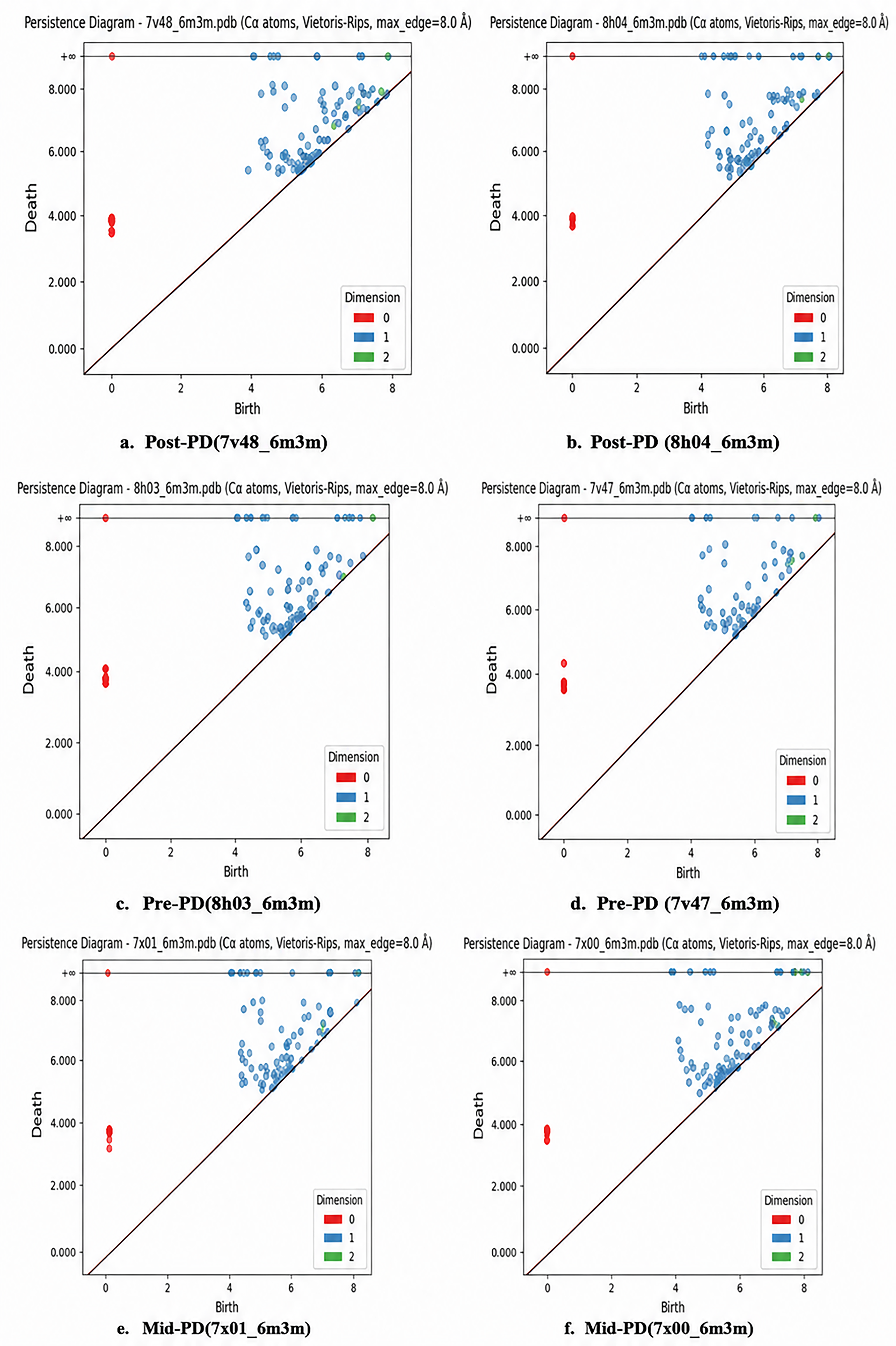
(a-f) Persistence diagram of docked complexes Post-PD, Pre-PD, and Mid-PDand 6m3m (SARS-CoV2 N-protein).
Maximum β-sheet content (max β0, β1, β2) in α-synuclein fibril polymorphs across Parkinson's disease stages following 6m3m docking.

Table 3 compares predicted binding-site residues (potential small-molecule pockets on α-syn fibrils) from TopoBind (a topology-informed method using persistent homology features such as max β0/β1/β2, SASA, and ESM-2 embeddings) and PyRosetta (a physics/energy-based Rosetta tool for pocket detection and refinement) across patient-seeded α-syn fibril structures from different PD stages (Pre-PD: 7v47_6m3m and 8h03_6m3m; Mid-PD: 7xo0_6m3m and 7xo1_6m3m; Late-PD: 8h04_6m3m and 7v48_6m3m). For each structure, it lists predicted residue ranges (as chain IDs, typically sequential segments in the fibril protofilaments) for chain A and chain B (the two protofilaments in the asymmetric fibril unit), the total number of predicted residues per method, and overlap metrics (absolute count and percentage) when aligning TopoBind predictions to PyRosetta's (and vice versa).
Summary of TopoBind and PyRosetta interface residue predictions and overlap percentages.
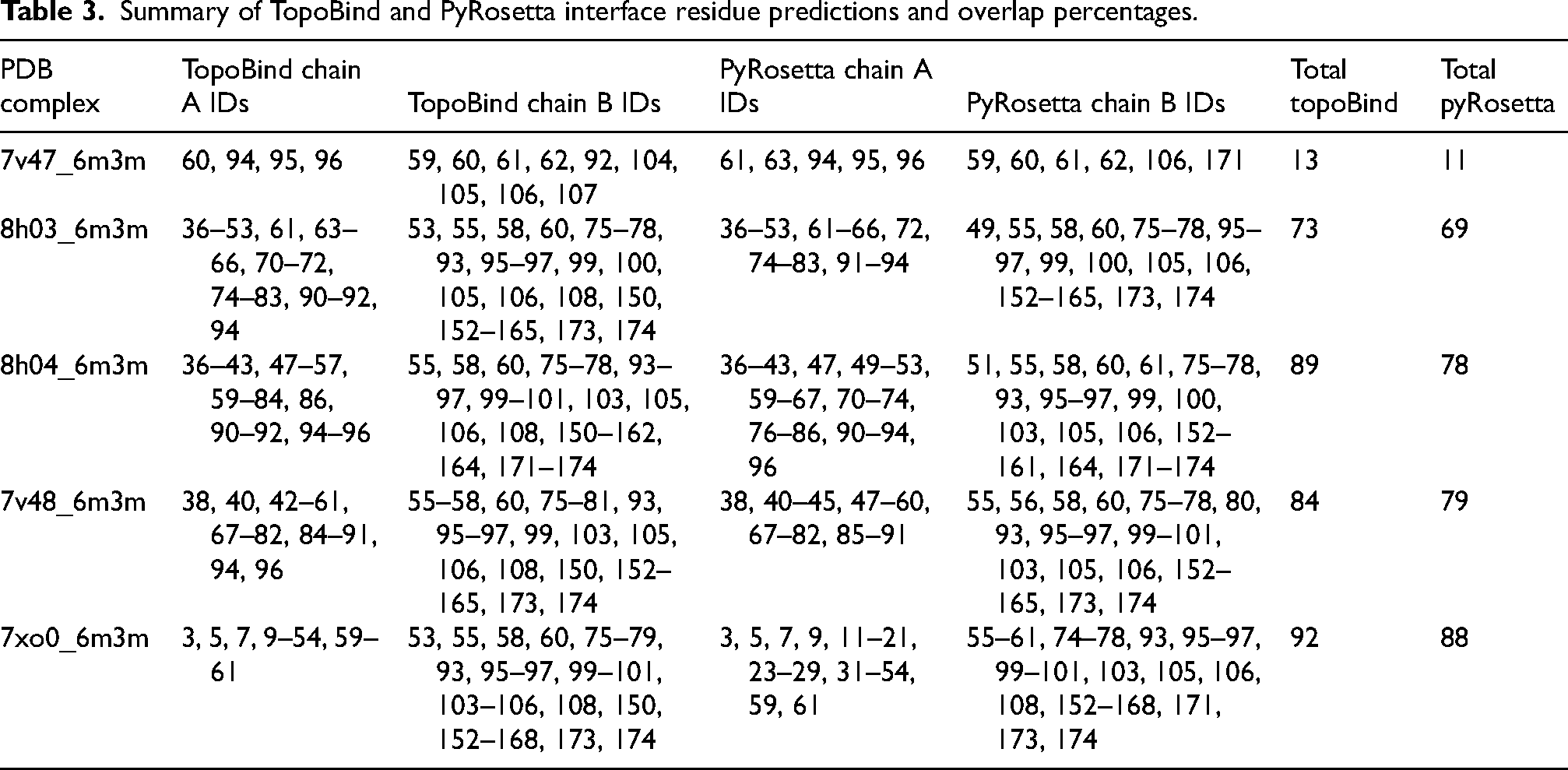
Overall, high overlap is observed (typically 74–92% in one direction, 55–91% in the other), with strong concordance across most structures (>80–92% in mid-PD and several late-PD cases), indicating that both methods identify largely the same residue clusters as druggable pockets despite methodological differences—TopoBind excels at capturing multi-scale topological loop/void patterns (linked to maturing β-sheet features), while PyRosetta emphasizes energetic favorability and sampling. Pre-PD shows more variability (e.g., lower 55–75% in 7v47, high 87–92% in 8h03), likely due to greater polymorphism/disorder in early aggregates, leading to less stable pockets. Mid-PD exhibits consistently high agreement (87–92%), suggesting stabilization of binding hotspots as fibrils mature. Late-PD maintains robust overlap (67–91%), with mature cross-β cores and interfaces yielding well-defined, conserved pockets.
These results indicate that TopoBind and PyRosetta converge on reliable, high-confidence binding sites—often in the NAC domain (residues ∼61–95), pre-NAC/grooves (e.g., around Y39, N65, G86, A85), or protofilament interfaces—common in α-syn fibrils for small-molecule binding (e.g., inhibitors that disrupt seeding/toxicity). The topology-driven TopoBind likely benefits from prior persistent homology trends (rising max β1 in later stages), improving detection in flexible/polymorphic systems. Overlapping residues represent prioritized therapeutic targets for fibril-disrupting compounds in PD, with stage-dependent consistency supporting the maturation of druggable features in advanced disease.
The docking analysis of the SARS-CoV2 N-protein 6m3m against various α-syn fibril polymorphs reveals a striking pattern of progressively increasing binding affinity as PD advances through its clinical stages. In fibrils representative of the preclinical or healthy-like (pre-PD) phase, such as those derived from structures 7V47 and 8H03, the predicted binding free energy (ΔG) ranges from approximately −8 to −13.8 kcal/mol, corresponding to dissociation constants (Kd) at 25°C between roughly 100 nM and 80 pM, as shown in Table 4. These modest values indicate weak to moderate interaction strength, suggesting that 6m3m has limited capacity to engage early-stage fibril conformations effectively, which may reflect less favorable binding pockets or lower surface complementarity in pre-pathological assemblies. Moving to mid-stage PD (mid-PD) fibrils, exemplified by 7X00 and 7X01, the binding becomes markedly stronger, with ΔG values shifting to the −15 to −18 kcal/mol range and predicted Kd estimates falling into the 100–500 pM regime. This transition from strong to very strong affinity implies that mid-stage fibril polymorphs begin to present more optimal structural features—potentially deeper or more complementary pockets—for 6m3m recognition, which could start to influence fibril stability, elongation, or templating behavior. The most dramatic enhancement occurs in late-stage, postmortem brain-derived (late-PD) fibrils, as seen in structures 8H04 and 7V48, where ΔG reaches −18 to −21.2 kcal/mol and predicted Kd values drop sharply into the femtomolar range (5–50 fM). These ultra-strong affinities indicate exceptionally tight and thermodynamically favorable binding, likely arising from highly evolved fibril conformations in advanced disease that create near-ideal interaction environments for the N-protein. Collectively, these results demonstrate a clear disease-stage-dependent increase in 6m3m binding potency, from weak in preclinical fibrils to ultra-strong in advanced pathological assemblies. This progressive enhancement suggests that α-syn fibril polymorphism evolves during PD progression in a manner that gradually improves N-protein accessibility and complementarity, raising intriguing possibilities for stage-specific therapeutic intervention—such as preferentially targeting mature, highly pathogenic fibrils in symptomatic patients—or for the development of conformation-selective diagnostic probes that exploit these structural differences across the disease spectrum.
Binding affinity of docked complex using prodigy.

PyRosetta-based binding affinity results show substantial variation in predicted binding strength across fibril polymorphs, even within the same clinical stage (e.g., pre-PD structures differ dramatically). The strongest interactions (most negative ΔG, largest ΔSASA) occur in mid- to late-stage (Mid-PD and Late-PD) fibrils, with ΔG values often ranging from −90 to −113 REU and ΔSASA > 3300 Å2, indicating highly favorable, extensive interfaces. In contrast, some pre-PD fibrils exhibit much weaker binding (e.g., 7v47), as shown in Table 5. These findings suggest that α-syn fibril structural polymorphism evolves during PD progression, creating more complementary and energetically favorable binding sites for 6m3m, particularly in mature, pathological assemblies from PD brain tissue.
Binding affinity of docked complexed using PyRosetta.

Table 6 summarizes the most energetically favorable residue contributors across multiple fibril polymorph complexes, based on per-residue total energy decomposition in Rosetta Energy Units (REU). Each row lists a residue identified as a key stabilizer of the fibril interface, including the complex name, residue number, chain identifier, amino acid type, and its computed total energy (more negative values indicate stronger favorable contributions). Across the analyzed polymorphs, the procedure consistently highlighted 10–40 significant residues per complex, with the top contributors typically ranging from −2.0 to −6.5 REU (and occasionally lower). Proline (PRO), leucine (LEU), phenylalanine (PHE), and tyrosine (TYR) frequently appear among the strongest contributors, reflecting their critical roles in backbone conformational constraint, hydrophobic packing, and potential π-stacking or steric interactions in the fibril core. The recurrence of certain residue types and energy profiles across different complexes underscores conserved energetic hot spots that drive fibril stability and polymorphism, providing valuable insights into the molecular determinants of amyloid assembly and potential targets for therapeutic modulation.
The PRODIGY-derived interaction profile for the docked complexes of 6m3m with α-syn fibril polymorphs across PD stages provides a detailed breakdown of intermolecular contact types and non-interacting surface (NIS) contributions, offering mechanistic insight into the observed binding trends. In the pre-PD fibril complexes (7V47_6m3m and 8H03_6m3m), the interaction landscape varies considerably. The 7V47 complex exhibits a relatively balanced but low-contact profile (4 charged–charged, 3 charged–polar, 6 charged–apolar, 2 apolar–apolar) with a high NIS apolar fraction (50.0%), consistent with limited interface complementarity and weak overall binding, as shown in table 7. In contrast, 8H03_6m3m shows a much richer interaction network (2 charged–charged, 7 charged–polar, 25 charged–apolar, 5 polar–polar, 36 polar–apolar, 39 apolar–apolar), with substantial contributions from apolar–apolar (39.0) and polar–apolar (36.0) contacts and a still-high NIS apolar component (50.0%), indicating that this pre-PD polymorph presents a more hydrophobic and extended interface capable of supporting very strong binding. Mid-PD complexes (7X00_6m3m and 7X01_6m3m) display further maturation of the interface, with increased charged–apolar (20–28), polar–apolar (40–48), and apolar–apolar (40–48) interactions, accompanied by a gradual decrease in NIS apolar fraction (49.0 to 48.5%), suggesting progressive optimization of hydrophobic and mixed-polar contacts that contribute to the transition from moderate to extremely strong affinity. Late-PD brain-derived fibrils (7V48_6m3m and 8H04_6m3m) exhibit the most extensive and diverse interaction profiles, characterized by elevated charged–apolar (17–33), polar–apolar (44–47), and apolar–apolar (42–46) contributions, along with relatively high charged–charged (5–7) and polar–polar (5–9) interactions and modestly reduced NIS apolar values (48.19–47.8%). These patterns indicate that mature pathological fibril conformations create larger, more chemically diverse interfaces with enhanced hydrophobic burial and balanced electrostatic/polar contributions, which collectively drive the ultra-strong binding observed in late-stage assemblies. Overall, the PRODIGY analysis underscores a disease-stage-dependent evolution of the 6m3m–fibril interface, progressing from limited and predominantly hydrophobic contacts in early fibrils to highly complementary, multi-type interaction networks in advanced late-PD structures, providing a molecular basis for the progressively increasing binding potency and supporting the hypothesis that fibril polymorphism during PD progression generates increasingly favorable environments for small-molecule engagement.
Top residue contributors ranked by total energy (REU) per complex.

Quantitative analysis of contact types and exposed surface contributions in 6m3m–α-synuclein fibril interfaces across pd progression stages.

In brief, this study harnesses persistent homology to quantitatively characterize the topological maturation of patient-derived α-syn fibrils pre-PD, mid-PD, and late-PD phases, using cryo-EM conformers such as 8H03 and 7V47 (pre-PD), 7XO0 and 7XO1 (mid-PD), and 8H04 and 7V48 (late-PD). The maximum persistent Betti numbers reveal progressive structural ordering: modest compaction via rising max β0 (186–187 in pre-PD to 188–189 in late-PD), a dominant increase in 1D loop/cycle persistence (max β1 from 63–81 in pre-PD to 81–85 in late-PD) linked to stabilized β-sheet ladders and cross-β twists, and minor max β2 rises indicating emerging peripheral pockets in mature fibrils. These multi-scale topological signatures provide interpretable evidence of fibril rigidification, reduced polymorphism, and enhanced pathogenic potential in advanced PD, aligning with cryo-EM conformational shifts and seeding studies.
These persistent homology features, fused with SASA and ESM-2 embeddings in a multi-modal pipeline (TopoBind-like), enabled reliable prediction of protein-protein interface residues (e.g., in α-syn/SARS-CoV-2 N-protein interactions) on α-syn fibrils, with high overlap (74–92%) against PyRosetta predictions confirming conserved hotspots in NAC grooves, protofilament interfaces, and hydrophobic regions.
Docking validations reinforce maturation-dependent affinity gains: Prodigy shows progression from very weak/strong in pre-PD (ΔG = -4.8 to −13.8 kcal/mol, Kd ≈ 2.9 μM to 80 pM) to strong/very strong in Mid-PD (ΔG = -15 to −18 kcal/mol, Kd ≈ 100–800 pM) and ultra-strong in late-PD (ΔG = -15.2 to −19.5 kcal/mol, Kd down to femtomolar levels). PyRosetta docking yields more negative dG(REU) in mid- and late-PD cases (−91.2 to −113.0 REU, SASA 2893–3781 Å2) than in weaker pre-PD cases, correlating with larger interfaces and stronger binding in mature fibrils.
The results from the ANM cross-correlation maps, computed using ProDy 27 on the multi-chain cryo-EM structures of CSF-amplified α-syn fibrils (PDB codes 7V47, 8H03, 7V48, 8H04, 7XO1, and 7XO0), reveal a clear stage-dependent shift in inter-protofilament dynamical coupling that complements and extends the structural and pathological findings. 21 In preclinical or healthy-like samples (7V47 and 8H03, dominated by Type 1A polymorph), the correlation matrices display prominent anti-correlated regions (deep blue) across chain boundaries, particularly in off-diagonal blocks and crossing the yellow lines marking approximate monomer interfaces (∼140 residues), indicating out-of-phase collective motions such as opposing twisting or shear between protofilaments as shown in the Figure 4(a) and 4(b). This dynamical decoupling aligns with the looser inter-protofilament packing and flexible N-terminal regions characteristic of Type 1A fibrils, which exhibit lower seeding potency in neuronal assays. In contrast, late-stage fibrils (7V48 and 8H04, enriched in Type 1B with contributions from Type 3) show markedly stronger and more extended positive correlations (intense red bands) spanning distant residue indices and crossing chain boundaries, reflecting enhanced in-phase, coherent motion across protofilaments—likely driven by tighter interface packing, altered helical twist, and the emergence of conformational subpopulations that strengthen harmonic connections in the elastic network as shown in the Figure 4(c) and 4(d). Mid-stage samples (7XO1 and 7XO0) exhibit intermediate patterns, with emerging but less uniform cross-coupling as shown in the Figure 4(e) and 4(f) are consistent with the gradual compositional shift toward pathogenic conformers described in the original study. 21 These dynamical differences provide a mechanical explanation for the paper's key observation that late-PD fibrils are significantly more potent at inducing endogenous α-syn aggregation and pathology: the increased inter-chain coherence may confer greater mechanical stability, reduce dissipative internal motions, and present more uniform templating surfaces, thereby enhancing persistence, resistance to fragmentation, and seeding efficiency during disease progression.

(a-f) Cross-correlation motionc(ANM) for docked complexes (Pre-PD, Post-PD, and Mid-PD) fibrils and SAR-Cov2-N protein (6m3m).
Discussion
Notably, these findings gain mechanistic relevance given that SARS-CoV-2 accelerates PD progression. Emerging evidence indicates that SARS-CoV-2 infection—particularly via direct interaction of its N protein with α-syn—accelerates amyloid aggregation and fibril formation in vitro and exacerbates α-syn pathology, dopaminergic neuron toxicity, neuroinflammation, and cellular phenotypes in human dopaminergic models and mouse systems pre-exposed to preformed fibrils. This viral promotion of α-syn misfolding/aggregation, alongside sustained neuroinflammation, oxidative stress, and potential brainstem involvement, may unmask or hasten PD onset/Parkinsonism in susceptible individuals and worsen symptoms or progression in those with preexisting disease. Our topology-informed insights into fibril maturation and enhanced binding interfaces in later stages thus highlight a plausible pathway by which SARS-CoV-2 could amplify pathogenic α-syn conformers, supporting long-term neurological risks post-infection. Overall, persistent homology emerges as a powerful descriptor for staging fibril pathology and uncovering disease-relevant evolution. The integrated prediction and docking framework advances computational strategies for identifying maturation-specific targets, paving the way for small-molecule or peptide modulators to disrupt seeding, elongation, or neurotoxicity in PD. The above results are shown in Figure 5, which provides a schematic illustration that integrates structural evolution, computational topology, binding site prediction, and viral interaction to explain pathogenic α-syn fibrillization. (A) Evolution of α-syn fibril structure: α-syn transitions from early, loosely organized fibrils to dense, β-sheet–rich mature fibrils under normal aggregation conditions. When SARS-CoV-2–related factors are present, fibril maturation speeds up, producing denser and more topologically complex fibrillar assemblies. (B) Persistent homology analysis: Topological barcodes and persistence diagrams display multiscale topological features (H0: connected components, H1: loops, H2: voids) that develop during fibril maturation. Mature and SARS-CoV-2–accelerated fibrils show longer-lasting and higher-order topological features, indicating increased structural stability and complexity compared to early fibrils. (C) Binding site prediction: Computational mapping reveals limited, spatially confined binding sites on early fibrils, while accelerated fibrils exhibit more and diverse surface-accessible binding pockets, suggesting greater interaction potential and toxicity. (D) SARS-CoV-2 interaction: The viral spike protein is proposed to act as a heterologous nucleation or scaffolding factor, promoting rapid α-syn aggregation and fibril growth. Overall, Figure 5 highlights that topological maturation, measured by persistent homology, along with changes in binding landscapes and viral influence, contribute to increased fibril stability, aggregation propensity, and pathogenicity in PD.

Topological development of α-synuclein fibrils in Parkinson's disease and acceleration by SARS-CoV-2.
Our novel hypothesis
Figure 5 illustrates a multiscale conceptual framework that integrates structural biology, computational topology, and viral interaction biology to explain how α-syn fibrils mature into pathogenic forms. The schematic is divided into four interconnected modules: fibril structure evolution, persistent homology analysis, binding site prediction, and SARS-CoV-2–mediated acceleration of aggregation. Together, these elements demonstrate how structural complexity, topological features, and external biological stressors collaborate to promote fibril maturation and increase toxicity
The first panel, α-syn fibril structure evolution, illustrates the step-by-step transformation from initial fibrillar assemblies to mature fibrils and ultimately to SARS-CoV-2–accelerated fibrillar forms. In the initial stage, α-syn aggregates appear loosely packed, structurally diverse, and less complex in topology. These early fibrils feature dynamic conformations with flexible loops and partially organized β-sheet structures. As aggregation continues under normal physiological conditions, intermolecular β-sheet stacking increases, leading to more compact, ordered, and thermodynamically stable mature fibrils. The structural development involves increased intermolecular hydrogen bonding, reinforcement of the hydrophobic core, and intertwining of protofilaments. The SARS-CoV-2–accelerated fibril represents a more intense state of aggregation, characterized by denser packing, altered surface topology, and possibly greater polymorphism. This indicates that viral-associated molecular interactions may reduce the nucleation barrier, boost seeding efficiency, or stabilize aggregation-prone conformations, thereby shortening the lag phase of fibrillization and encouraging rapid structural maturation.
The second panel introduces persistent homology analysis, a computational topology framework that quantifies structural complexity beyond traditional geometric descriptors. Persistent homology captures the birth and death of topological features—such as connected components (H0), loops (H1), and voids (H2)—across multiple spatial scales. The topological barcode representation shows how these features persist as the filtration parameter increases, reflecting the structural robustness of fibrillar organization. In early fibrils, fewer long-lived topological features are observed, indicating limited structural integration. As fibrils mature, additional persistent loops and cavities emerge, representing stabilized intermolecular arrangements and internal structural heterogeneity. The persistence diagram further visualizes the lifetime of these features, with longer-lived points corresponding to highly stable structural motifs. SARS-CoV-2–accelerated fibrils may exhibit altered persistence features, potentially showing enhanced H1 and H2 features that reflect tighter packing and increased cross-sectional complexity. This approach provides a quantitative metric for distinguishing fibril polymorphs and assessing maturation states, offering a mathematically rigorous description of aggregation progression.
The third panel highlights binding site prediction, focusing on how surface topology changes during fibril maturation. Early fibrils have limited, partially exposed binding sites with moderate electrostatic and hydrophobic complementarity. Computational mapping indicates that as fibrils mature, the number and distribution of accessible pockets increase, especially at protofilament interfaces and surface grooves. These new pockets may improve interactions with cellular proteins, membranes, metal ions, or inflammatory mediators. In SARS-CoV-2–accelerated fibrils, altered surface curvature and electrostatic properties may further expand or modify these binding regions. Such changes could increase cytotoxic potential by promoting abnormal protein–protein interactions, membrane permeabilization, or immune activation. Higher binding-site density is associated with greater aggregation propensity and toxicity, linking structural maturation to pathological outcomes.
The fourth panel shows SARS-CoV-2 interactions, specifically suggesting that viral spike protein interactions may accelerate α-syn aggregation. Viral proteins could act as heterogeneous nucleation surfaces, providing scaffolds that stabilize aggregation-prone shapes. Electrostatic complementarity between spike protein components and α-syn monomers or oligomers might increase local concentration and promote conformational changes toward β-sheet–rich structures. This interaction could also cause conformational strain or structural rearrangements, leading to fibril polymorphs that differ from those formed under normal conditions. The faster aggregation pathway implies that viral infection or inflammatory responses could influence neurodegenerative processes by changing protein misfolding rates. Although mechanistic details need experimental validation, the model offers a plausible biophysical pathway linking viral exposure to altered fibril growth.
The integrated model shown in Figure 5 highlights that fibril maturation is not merely an increase in aggregate size but a topological transformation involving multiscale structural reorganization. Persistent homology offers a robust analytical framework for quantitatively capturing these changes, linking structural biology and computational mathematics. Binding-site evolution connects structural maturation to functional and pathological outcomes, while viral interaction introduces an external modulatory factor that can alter aggregation pathways.
Importantly, Figure 5 shows that improved fibril maturation is associated with greater toxicity and disease relevance. As structural complexity and binding interface density grow, fibrils may become more resistant to proteolytic clearance, more effective in templated seeding, and more disruptive to cellular homeostasis. The topological maturation framework thus provides a unified view where structural, mathematical, and biological aspects come together to explain disease progression.
In summary, this schematic suggests that α-syn aggregation in PD can be viewed as a progressive topological development, measured through persistent homology, interpreted through binding-site prediction, and possibly influenced by viral factors such as SARS-CoV-2. By combining computational topology with structural and molecular insights, the model promotes a systems-level understanding of fibril maturation and explores opportunities for quantitative biomarkers, therapeutic targeting of aggregation interfaces, and the study of infection-related neurodegenerative acceleration.
Footnotes
Acknowledgements
All the authors thank their Institutions for their support.
Ethical considerations
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We gratefully acknowledge the funding support received from the CERD PhD Research Fellowship Programme of APJ Abdul Kalam Technological University under Order No. 662/2022/KTU to Pranathi Jalapally and KLEF-2 Doctoral funding to Lakshmi Sowmya Emani. This work was supported by the KLEF, (grant number 002).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data generated or analyzed during this study are included in this published article and available from the corresponding author's laboratory.