
Abstract
CRISPR activation (CRISPRa) offers a powerful approach to upregulate endogenous genes; yet, existing systems in plants can be complex or difficult to integrate with CRISPR interference (CRISPRi). Here, we present a streamlined and flexible CRISPRa platform that enables robust gene activation. Using a dual-luciferase reporter, we benchmarked a range of guide RNA scaffolds, effector proteins, and promoters. We developed a novel single-guide RNA (sgRNA) architecture, harboring two MS2 aptamers inserted into the tetraloop and driven by a composite Pol II/Pol III promoter, as the most efficient configuration. This scaffold outperformed gR2.0- and SunTag-based constructs, reaching up to 100-fold activation of a minimal 35S promoter and up to 215-fold induction of three endogenous rice genes in protoplast assays. In contrast, scaffold RNAs (scRNAs) with aptamers at the 3′ end or in excessive copy numbers were ineffective. Exploratory AlphaFold modeling supports a possible role for aptamer positioning and MCP-VP64 dimerization, although this remains a working hypothesis. This modular design enables tunable gene regulation in rice protoplasts and provides a practical platform for high-throughput screening and synthetic gene circuit prototyping in plants. Given that scRNA geometry and promoter architecture are universal features of CRISPR-based transcriptional modulation, the system is expected to be broadly portable across species. While the architecture is intended to be compatible with CRISPRi, future studies will be needed to establish its practical use in combined CRISPRa/i settings.
Introduction
CRISPR activation (CRISPRa) enables simultaneous upregulation of multiple genes, a task that remains challenging with conventional overexpression approaches. As a result, CRISPRa has attracted growing interest in plant research. For further details, see.1–4 While highly effective CRISPRa systems exist in plants, their complexity and lack of compatibility with CRISPR interference (CRISPRi) limit broader applications.5,6 This highlights the need for a simplified and portable architecture that maintains high activation efficiency while being easily adaptable across species and compatible with CRISPRi frameworks.
Initial plant CRISPRa systems used dCas9 fused to VP64 or EDLL, yielding up to 12-fold activation.7,8 A key step toward improving activation efficiency has been the identification of alternative transcriptional activators. Notably, the dCas9-TV construct, combining a 6xTALE activation domain from Xanthomonas with two VP64 units, outperforms earlier fusions in both plant and mammalian cells.9,10 Beyond current constructs, CRISPRa performance can be further enhanced by exploring novel transcriptional activator domains. Recent screens of plant transcription factor domains identified activators that surpass VP16, a long-standing benchmark.11,12
Beyond standard CRISPRa strategies, activation levels can be further enhanced through the use of modular RNA scaffolds (scRNA). By combining dCas9-VP64 and an scRNA carrying two MS2 aptamers recruiting additional copies of the VP64 protein, Lowder et al. developed the Act2.0 system. 13 Selma et al. created an improved scRNA (scRNA2.1) by inserting a nucleotide into the loop of the MS2 aptamer. 14 Another key strategy relies on activator multimerization, notably via the SunTag system or the MoonTag, which enhances recruitment efficiency and stability. In the SunTag system, 10 GCN4 peptide repeats are fused to dCas9, enabling the recruitment of 10 VP64 domains via ScFv antibodies that specifically recognize the GCN4 epitope. 15 MoonTag retains the multimerization logic of SunTag, but its components are different and optimized for enhanced expression and stability in plant systems, resulting in improved overall efficiency. 16
Act3.0 and MoonTag currently represent the most advanced CRISPRa platform in plants, combining aptamer-guided recruitment and SunTag/MoonTag amplification to achieve strong transcriptional output.5,16 The tripartite design of Act3.0 combines (i) a dCas9-VP64 fusion; (ii) a gR2.0 scaffold bearing two MS2 aptamers; and (iii) a SunTag array of 10 GCN4 peptides fused to MCP, which recruits a 2xTAD via ScFv. This system outperforms the second-best construct, dCas9-TV (6xTALE-VP128), by a factor of 4 to 6, and enables strong overactivation of target genes such as OsER1 and OsGW7 by 100- to 250-fold, respectively. 5 Its originality lies in the use of aptamer-guided SunTag recruitment, offering synergistic enhancement beyond dCas9 fusions alone. In this context, the gR2.0 scaffold also appears more effective than the scRNA2.1 scaffold, 14 at least within the Act3.0 configuration. Moreover, the system is amenable to multiplexing, further expanding its potential for complex gene network reprogramming in plants.
Dual CRISPRa/i systems repurpose dCas9 as a scaffold for modular recruitment of activators and repressors via RNA-guided aptamers. Here, dCas9 functions solely as a recruitment scaffold. Transcription activators and repressors are recruited uniquely by two different classes of scRNAs containing two types of viral protein-binding aptamers, such as MS2 and com. In this configuration, the corresponding viral protein MCP and Com are fused to two distinct types of transcription effectors. 17 Their work laid the groundwork for defining operational rules, including aptamer positioning, interference minimization, and scaffold stability. Aptamers positioned at the 5′ end of the scRNA appeared less efficient than those placed in the 3′ region. 17 Moreover, aptamer multimerization tends to reduce regulatory efficiency and expression. To address this, Zalatan et al. introduced a new scaffold design in which double-stranded linkers separate aptamer repeats, improving RNA stability and enabling simultaneous activation and repression of target genes (CXCR4 and B4GALNT1) in human cells 17 using VP64 and KRAB effectors. Notably, they successfully redirected the violacein biosynthetic pathway using this orthogonal regulatory framework. 17 This system is now used, for example, to reprogram cell differentiation in the biomedical field, such as, chondrogenesis in regenerative medicine. 18
Here, we introduce a streamlined CRISPRa system, validated in rice protoplasts, and an architecture intended to be broadly portable and compatible with CRISPRi. Using this system, we showed that the combination of a novel scRNA scaffold (2xMS2 inserted in the tetraloop), a composite Pol II/Pol III promoter, and MCP-VP64 significantly outperformed even advanced SunTag-based constructs for CRISPRa activation. We also performed AlphaFold simulations and provided a plausible hypothesis-driven structural explanation for the observed differences in scRNA efficiency, offering a rational framework for future scaffold optimization. Unlike existing systems requiring complex multicomponent assemblies, our design achieves high efficiency with minimal elements and provides a flexible, broadly applicable platform for future orthogonal CRISPRa/i applications across plant and potentially nonplant systems.
Experimental Procedures
Molecular biology
PCR amplification using KOD Xtreme™ Hot Start polymerase
PCR reactions were performed in a final volume of 50 µL, containing: 25 µL of 2x KOD Xtreme buffer, 10 µL of dNTP mix (2 mM each), 1.5 µL each of forward and reverse primers (10 µM), 1 µL of KOD Xtreme Hot Start DNA Polymerase (VWR ref 71975–3), and nuclease-free water to complete the volume. Thermal cycling conditions were as follows: initial denaturation at 98°C for 2 min, followed by 30 cycles of 98°C for 10 s, 62°C for 30 s, and 68°C for 1 min per kilobase of expected amplicon length.
Gel purification
PCR products or restriction-digested vectors were resolved on 1% agarose gels and purified using the Zymoclean™ Gel DNA Recovery Kit (Zymo Research), following the manufacturer’s instructions. DNA was eluted in a final volume of 12 µL of elution buffer or nuclease-free water. DNA concentration and purity were assessed using a NanoQuant spectrophotometer (Tecan).
In-Fusion cloning
Primers were designed using the Takara In-Fusion® online design tool (https://takara.teselagen.com/#/DesignPage), according to the manufacturer’s guidelines, and used for PCR amplification or gene synthesis with KOD Xtreme™ Hot Start DNA Polymerase. In-Fusion cloning reactions were performed following the supplier’s protocol (Takara™). Briefly, 2 µL of 5x In-Fusion HD Enzyme Premix was added to 150 ng of linearized vector and PCR products at a 2:1 insert-to-vector molar ratio. The mixture was incubated at 50°C for 15 min, then placed on ice and either stored at −20°C or immediately used for transformation. For bacterial transformation, 2.5 µL of the reaction mixture was added to 50 µL of Stellar™ competent cells (Takara™), according to the manufacturer’s instructions.
Gene synthesis
Primers for gene synthesis were designed using the Primerize web tool (https://primerize.stanford.edu) with the following parameters: Tm = 60°C and primer length between 60 and 80 nucleotides. Gene assembly was performed by mixing 1.5 µL of each primer in a PCR reaction using KOD Xtreme™ Hot Start DNA Polymerase. External primers were used at 100 µM and internal overlapping primers at 10 µM. PCR amplification was carried out as described above using the same cycling conditions.
Sequencing
Sanger sequencing primers were designed using the Genscript™ DNA Sequencing Primer Design Tool (https://www.genscript.com/tools/dna-sequencing-primer-design). Selected plasmids were also verified by whole plasmid nanopore sequencing, performed by Eurofins Genomics (https://eurofinsgenomics.eu/en/custom-dna-sequencing/eurofins-services/whole-plasmid-sequencing/).
CRISPRa constructions
The dual-luciferase cassette includes a Renilla luciferase (rLUC) gene, derived from a Promega vector, driven by a CaMV35S promoter, and a Firefly luciferase (fLUC) gene under the control of a tetO-responsive element fused to a minimal CaMV35S promoter and assembled by In-Fusion into dCas9-MCP-VP64.
The dCas9-MCP-VP64 plasmid was constructed by In-Fusion assembly of PCR-amplified fragments: (i) a dCas9 module under the control of the ZmUBI promoter, (ii) an MCP:VP64 module under the control of the CaMV35S promoter, and (iii) the dual-luciferase cassette (see Supplementary Table S1).
The SunTag plasmid was generated by a combination of PCR-based In-Fusion cloning and gene synthesis, using dCas9-MCP-VP64 as backbone. It includes: (i) a dCas9 module under the ZmUBI promoter, (ii) a MCP-(GCN4)10x module under the control of the CsCMV promoter, (iii) a ScFv:GFP:VP64:GB1 module under the CaMV35S promoter, and (iv) the dual-luciferase cassette (Supplementary Table S2) using Addgene#158408 as a template.
The Act3.0 plasmid was assembled using the SunTag vector as a backbone. It includes: (i) a dCas9:VP64 module under the ZmUBI promoter, (ii) a MCP-(GCN4)10x module fused translationally to dCas9:VP64 via a T2A peptide, (iii) a ScFv:GFP:VP64:GB1 module under the CaMV35S promoter, and (iv) the dual-luciferase cassette (Supplementary Table S1).
See also Supplementary Figure S3 and Supplementary Data.
scRNA constructions
Synthetic scRNA variants, including 5′ 2xMS2, 5′ 8xMS2, 3′ 2xMS2, and 3′ 8xMS2, were obtained from GenScript™ and cloned under the control of OsU3 promoters. 19 A synthetic composite promoter 20 was also synthetized (GenScript™). The 5′ 2xMS2 scRNA was PCR-amplified and inserted downstream of the composite promoter by In-Fusion cloning using BsaI/BsaI sites. Additional constructs (gR2.0 and 4xMS2) were similarly synthesized (GenScript™) and cloned into the composite promoter backbone using BsaI/BsaI sites. Spacers were designed for tetO, OsGW7, OsER1, and OsF3H and cloned by golden gate using BsaI/BsaI sites in empty scRNA. See Supplementary Table S2 for primers.
See also Supplementary Table S2, Supplementary Figure S1, and Supplementary Data.
Protoplast extraction and processing
Protoplasts were extracted from Kitaake Japonica rice following the protocol described 21 with minimal modifications. Briefly, after extraction, protoplast pellets were resuspended in 1 mL of W5 solution and incubated overnight in the dark at 25°C, with the tubes placed horizontally. The following day, tubes were centrifuged at 250 g for 10 min at room temperature, using reduced acceleration and deceleration settings (level 2). After supernatant removal, 500 µL of MMG solution was added to each pellet. Protoplast viability was assessed by FDA staining as described. 21 Mannitol – MgCl2 – MES (MMG) was added to adjust the concentration to a minimum of 1 × 106 protoplasts per mL. Tubes were then incubated for 1 h at 25°C in the dark. For transfection, 5 µg of plasmid DNA per construct was added to a single tube in a final volume not exceeding 20 µL. Then, 200 µL of protoplast suspension was added to each tube, followed by 200 µL of 40% PolyEthylene Glycol (PEG) solution. Tubes were gently mixed and incubated for 15 min in the dark at room temperature. The transfection reaction was stopped by adding 900 µL of W5 solution. Samples were centrifuged at 250 g for 10 min, the supernatant was removed, and 500 µL of fresh W5 solution was added to each pellet. Tubes were stored horizontally at 25°C in the dark for 72 h prior to analysis.
Protoplasts RNA extraction
Total RNA was extracted from rice protoplasts using Purelink RNA Mini Kit (Invitrogen Life Technologies), following the manufacturer’s protocol with minor modifications. Protoplasts were collected by centrifugation at 2000 g for 5 min at 4°C and immediately resuspended in 350 µL of lysis buffer (with β-mercaptoethanol). Samples were vortexed thoroughly and homogenized by pipetting. The lysate was transferred into a clean homogenization tube and centrifuged at 12,000 g for 2 min before proceeding with ethanol addition and binding to the spin cartridge. RNA was eluted in 30–40 µL of RNase-free water and quantified using a TapeStation. RNA was diluted in RNAse-free water at a final concentration of 2.5 ng/µL.
Luna® Universal One-Step RT-qPCR Protocol
Reverse transcription and quantitative PCR were performed using the Luna Universal One-Step RT-qPCR Kit (NEB, E3005), following the manufacturer’s instructions. Each 10 µL reaction contained: 5 µL Luna Universal One-Step Reaction Mix (2x), 0.5 µL Luna WarmStart® RT Enzyme Mix (20x), 0.4 µM forward primer, 0.4 µM reverse primer, 2 µL total RNA (5 ng), and nuclease-free water to 10 µL. Reactions were carried out in a Light cycle™ Real-Time PCR Detection System (Bio-Rad) using the following thermal profile: reverse transcription: 10 min at 55°C; initial denaturation: 1 min at 95°C; 45 cycles of: denaturation: 10 s at 95°C; annealing/extension: 30 s at 60°C (with plate read). Each sample was run in quadriplicate. Relative expression was normalized to the OsExp’ reference gene using the ΔΔCt method. Fold change was computed using CRISPRa control transformed with scRNA without any spacer.
Dual luciferase assays
After incubation, protoplasts were centrifuged for 1 min at 250 g, and the samples were transferred into 1.5 mL microcentrifuge tubes. A first centrifugation was performed at 2300 g for 10 min to pellet the cells, after which the W5 supernatant was carefully removed using a 1 mL pipette. A second brief centrifugation was carried out, and the remaining supernatant was removed using a 10 µL pipette to minimize residual buffer. Each pellet was then resuspended in 75 µL of 1x Passive Lysis Buffer (Promega). After vortexing and a short spin-down, lysates were transferred into white 96-well luminometer plates (Ref., 1173132279 Fisher Scientific). The Dual-Glo® Luciferase assay was performed according to the manufacturer’s instructions (Promega, Ref. E2920) using 75 µL of Dual-Glo® reagents per well. Luminescence was measured with a GloMax® Navigator plate reader (Promega) on four independent biological replicates.
Statistical analysis
GraphPad V10 was used for graph constructions and statistical analysis (student t-test).
Results
Design of a dual luciferase reporter to evaluate CRISPRa and scRNA efficiency
Our goal was to establish an optimized CRISPRa system in rice protoplasts, with an architecture intended to be compatible with CRISPRi, based on scRNA-mediated recruitment of transcriptional activators. To this end, we designed a dual luciferase reporter cassette to quantify activation efficiency across different CRISPRa and scRNA variants. The rLUC, driven by the constitutive p35S promoter, served as an internal control to normalize variations in transformation efficiency. The fLUC was placed under the control of a minimal p35S promoter fused to a modified tetO sequence derived from the tetracycline operator, containing a PAM site and targeted by a specific tetO spacer (Fig. 1A). Upon co-expression of the CRISPRa machinery and a matching scRNA targeting the tetO sequence, activation is triggered, and CRISPRa efficiency is quantified as the ratio of fLUC to rLUC luminescence (fLUC/rLUC), expressed in relative light units (Fig. 1A).
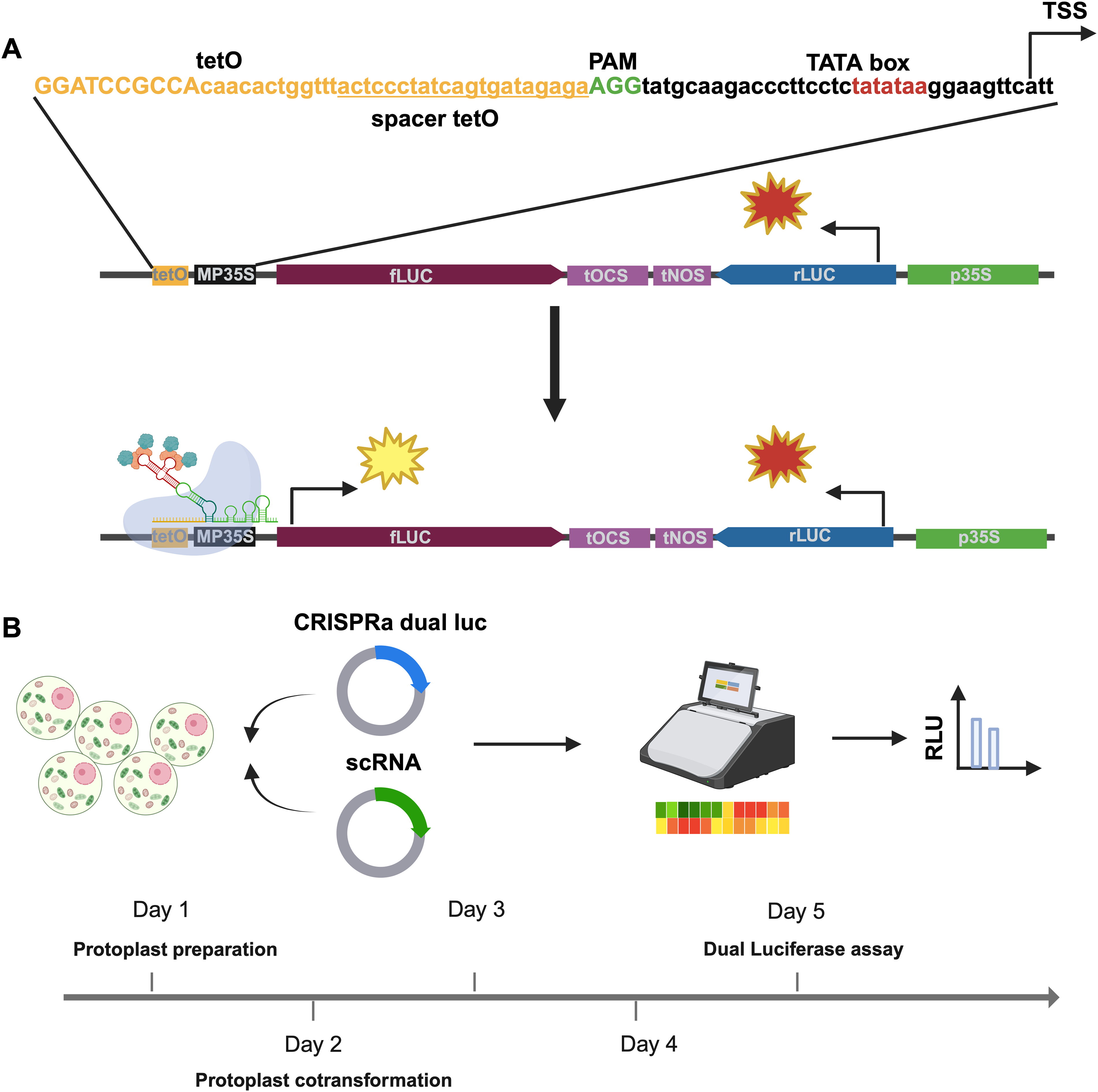
Dual luciferase system to evaluate CRISPRa constructs.
Development of a flexible and streamlined protoplast-based CRISPRa assay
To ensure flexibility, reproducibility, and compatibility with high-throughput screening, we established a modular protoplast transformation protocol in which protoplast isolation, transformation, and dual luciferase quantification are performed as separate steps (Fig. 1B). Protoplasts were first isolated from 10-day-old rice shoot seedlings on Day 1, then co-transformed on Day 2 with scRNA and CRISPRa constructs, and luciferase activity was measured on Day 5. This co-transformation approach significantly accelerates the testing pipeline by allowing rapid evaluation of multiple scRNA/CRISPRa combinations without requiring individual subcloning of scRNAs into CRISPRa vectors. In addition, decoupling the transformation from the isolation step simplifies logistics and increases throughput, enabling a single lab member to perform two independent transformation rounds per week. Finally, measuring luciferase activity after 72 h provides a more stable readout, corresponding to a plateau phase of expression levels compared with earlier time points, and thus improves the robustness and comparability of the data. Because the design is modular, the same workflow can, in principle, be adapted to other species or cell systems for rapid testing of CRISPRa/i components.
Comparison of CRISPRa efficiency across scRNA designs, effector constructs, and promoters
We designed a series of scRNAs containing single, double, or octuple MS2 aptamer insertions at the 3′ end, the tetraloop, and hairpin 2 regions (Fig. 2A; Supplementary Fig. S1), following the SIRIUS design framework. 23 The secondary structures of these scRNAs were predicted using RNAfolder, confirming low ΔG values and proper folding of the aptamer motifs (Supplementary Fig. S2). To evaluate CRISPRa performance, we tested an effector construct: dCas9-MCP-VP64 driven by pZmUBI (for dCas9) and p35S (for MCP-VP64) promoters (Fig. 2B; Supplementary Fig. S3). We also compared two strategies for scRNA expression: the U3 promoter (Pol III) and a composite promoter combining Pol II and Pol III elements (Fig. 2C; Supplementary Fig. S1), in order to optimize expression and CRISPRa efficiency.
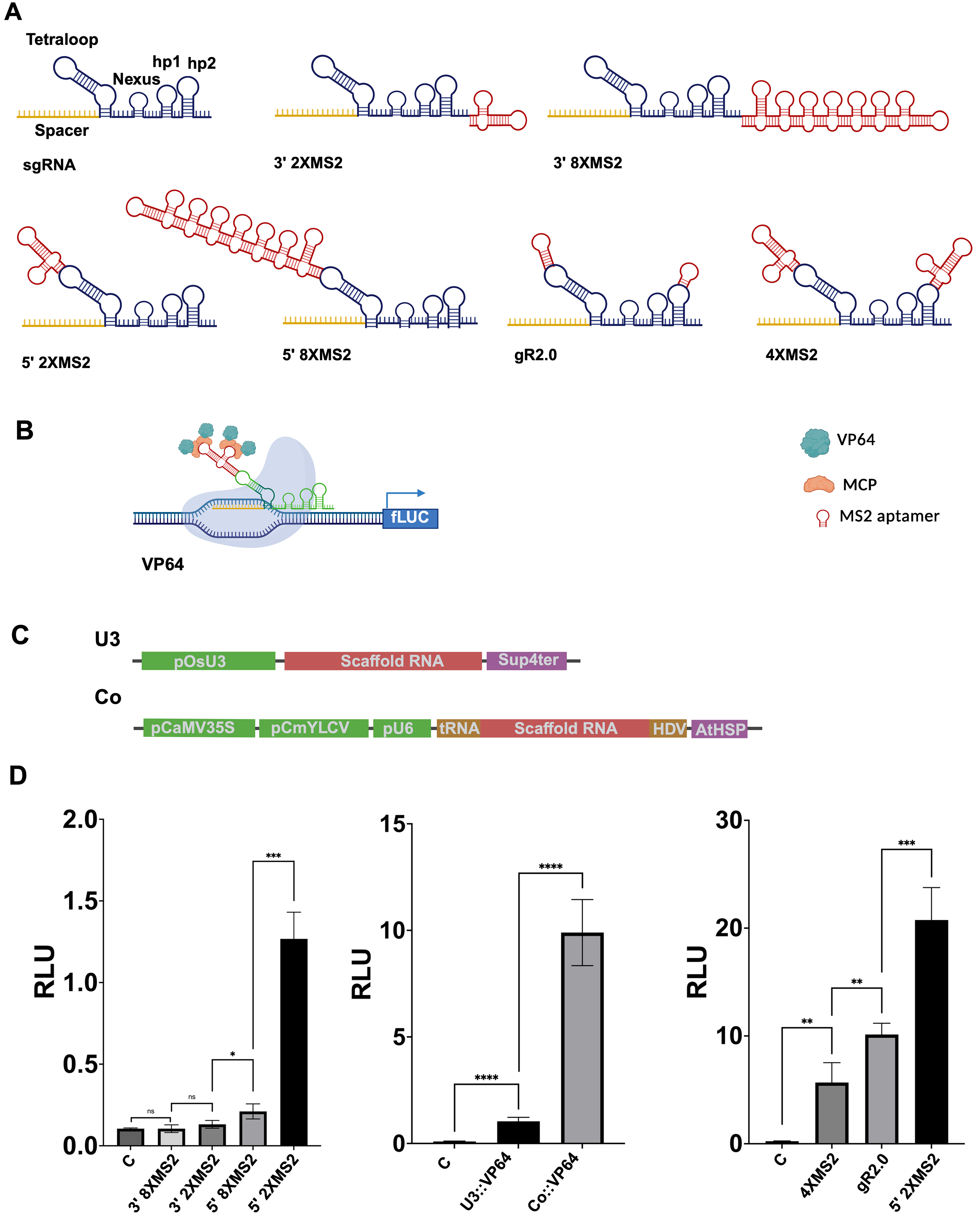
Comparison of CRISPRa effectors (VP64), scRNA designs, and U3 versus composite promoters for scRNA expression.
Effect of aptamer position and copy number on CRISPRa
We compared the activation efficiency of dCas9-MCP-VP64 using scRNAs carrying either double or octuple MS2 aptamer insertions at the 3′ end (3′-2xMS2, 3′-8xMS2) or within the tetraloop region (5′-2xMS2, 5′-8xMS2), all expressed under the U3 promoter (Fig. 2D, left panel). No activation of fLUC expression was observed with scRNAs containing aptamers in the 3′ end, regardless of copy number. In contrast, insertion within the tetraloop region significantly activated fLUC compared with the nontargeting control. Notably, the 2xMS2 tetraloop version induced a 6-fold higher activation than the 8xMS2 version, indicating that excessive aptamer copies may hinder efficiency.
Influence of scRNA promoter on CRISPRa efficiency
Use of a composite Pol II/Pol III promoter to drive scRNA expression was associated with 10-fold higher activation. These results suggest that differences in scRNA abundance or processing could contribute to the observed effects. However, scRNA levels were not quantified in this study, and additional work will be required to distinguish contributions of scRNA expression from effects driven by scaffold architecture (Fig. 2D, middle panel). Altogether, the combination of a composite scRNA promoter, dCas9-MCP-VP64, and 5′-2xMS2 scRNA achieves robust CRISPRa activity, reactivating the minimal p35S promoter by up to 100-fold in our system.
Benchmarking 5′-2xMS2 scRNA against gR2.0 and 4XMS2 variant
We next compared our best-performing construct, the 5′-2xMS2 scRNA, with the widely used gR2.0 scaffold from the Synergistic Activation Mediator (SAM) system, and to a newly designed 4xMS2 variant, which includes two additional MS2 aptamers inserted into hairpin 2, in an attempt to further enhance activation. All scRNAs were expressed under the composite promoter for consistency. In this configuration, 5′-2xMS2 outperformed both alternatives, showing a 2-fold increase in CRISPRa efficiency compared with gR2.0, which itself was twice as effective as the 4xMS2 variant (Fig. 2D, right).
Comparison of VP64 and SunTag-based CRISPRa constructs
To further enhance CRISPRa efficiency, we developed two new activation systems based on the SunTag strategy. The first construct incorporates two expression cassettes: one encoding MCP-(GCN4)10x under the control of the pCsCMV promoter and the other encoding ScFv-VP64 under p35S promoter (see Supplementary Fig. S3). In theory, this configuration enables the recruitment of up to 40 VP64 domains using a 5′ 2xMS2-modified scRNA (Fig. 3A). This architecture is, in principle, compatible with CRISPRi. Additionally, we designed a second construct, functionally similar to the Act3.0 system, where dCas9 is transcriptionally fused to VP64, and MCP-(GCN4)10x is co-expressed under the same pZmUBI promoter. The two proteins are separated by a T2A peptide to allow independent translation (Fig. 3A and Supplementary Fig. S3). Unlike the first system, Act3.0 is not compatible with CRISPRi as dCas9 is fused to a transcriptional activator.
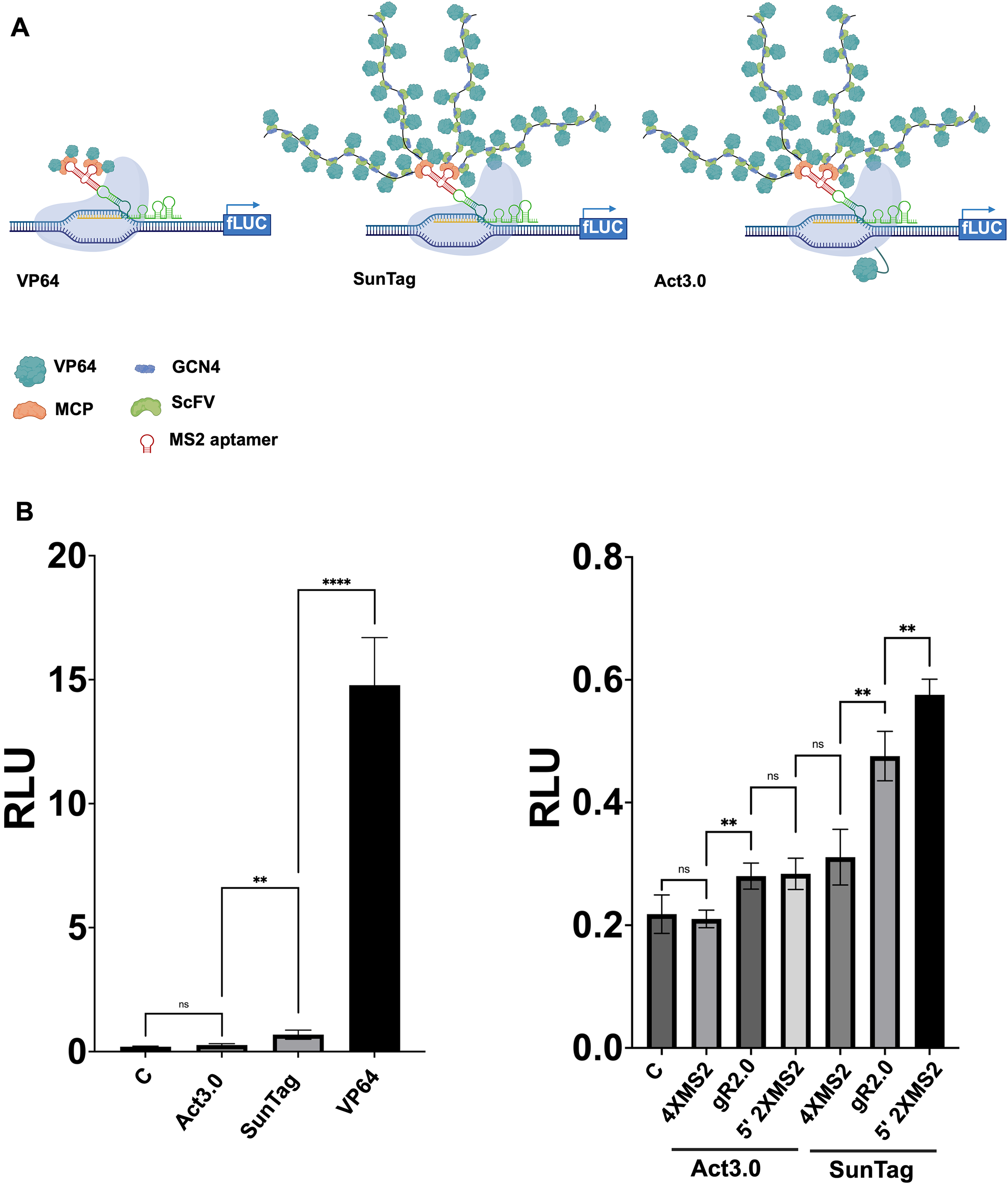
Comparison of CRISPRa effectors (VP64, SunTag, Act3.0) and scRNA designs used with SunTag and Act3.0 systems.
Unexpectedly, Act3.0 weakly induced measurable fLUC activation, while the SunTag-based construct yielded a more robust and significant activation. In comparison, MCP-VP64 activated fLUC expression with a 20-fold higher efficiency than the SunTag construct (Fig. 3B, left panel). As this experiment used the 5′ 2xMS2 scRNA, we hypothesized that the limited performance of the SunTag system might result from suboptimal guide RNA architecture. To test this, we compared the activation efficiency of both Act3.0 and SunTag constructs using three different scRNA configurations: 4xMS2, gR2.0, and 5′ 2xMS2 (Fig. 3B, right panel). Results confirmed that Act3.0 remained a poor activator across all configurations. In contrast, SunTag consistently outperformed Act3.0, regardless of the scRNA used. Notably, the 5′ 2xMS2 scRNA emerged as the most effective configuration for the SunTag system as for dCas9-MCP-VP64 construct.
CRISPRa on endogenous rice genes
To validate our most effective CRISPRa system, we selected three endogenous rice genes previously used to benchmark Act3.0-based SunTag constructs. 5 We employed the same guide RNA sequences described in that study to target the promoters of OsF3H, OsER1, and OsGW7 and assessed gene activation via one-step RT-qPCR (Fig. 4A). In our experimental system, both SunTag and MCP-VP64-based CRISPRa induced activation of OsF3H, with fold changes of approximately 7 and 215, respectively (Fig. 4A, left panel). For OsER1, activation levels were 2.7-fold (SunTag) and 34-fold (MCP-VP64) and for OsGW7, 2-fold and 12.5-fold (Fig. 4A, middle and right panels). These results confirm that the dCas9-MCP-VP64 system robustly activates transcription of endogenous rice genes and consistently outperforms the SunTag construct across all tested targets. Importantly, the high efficiency obtained with minimal components suggests that this architecture should be broadly transferable beyond rice, providing a flexible baseline for CRISPRa/i applications in diverse plant systems. We also compared the basal expression levels of these three genes, normalized to OsGW7 (Fig. 4B). Interestingly, the magnitude of CRISPRa-induced activation appeared negatively correlated with the basal expression level of each gene, a classical feature of CRISPRa experiments.
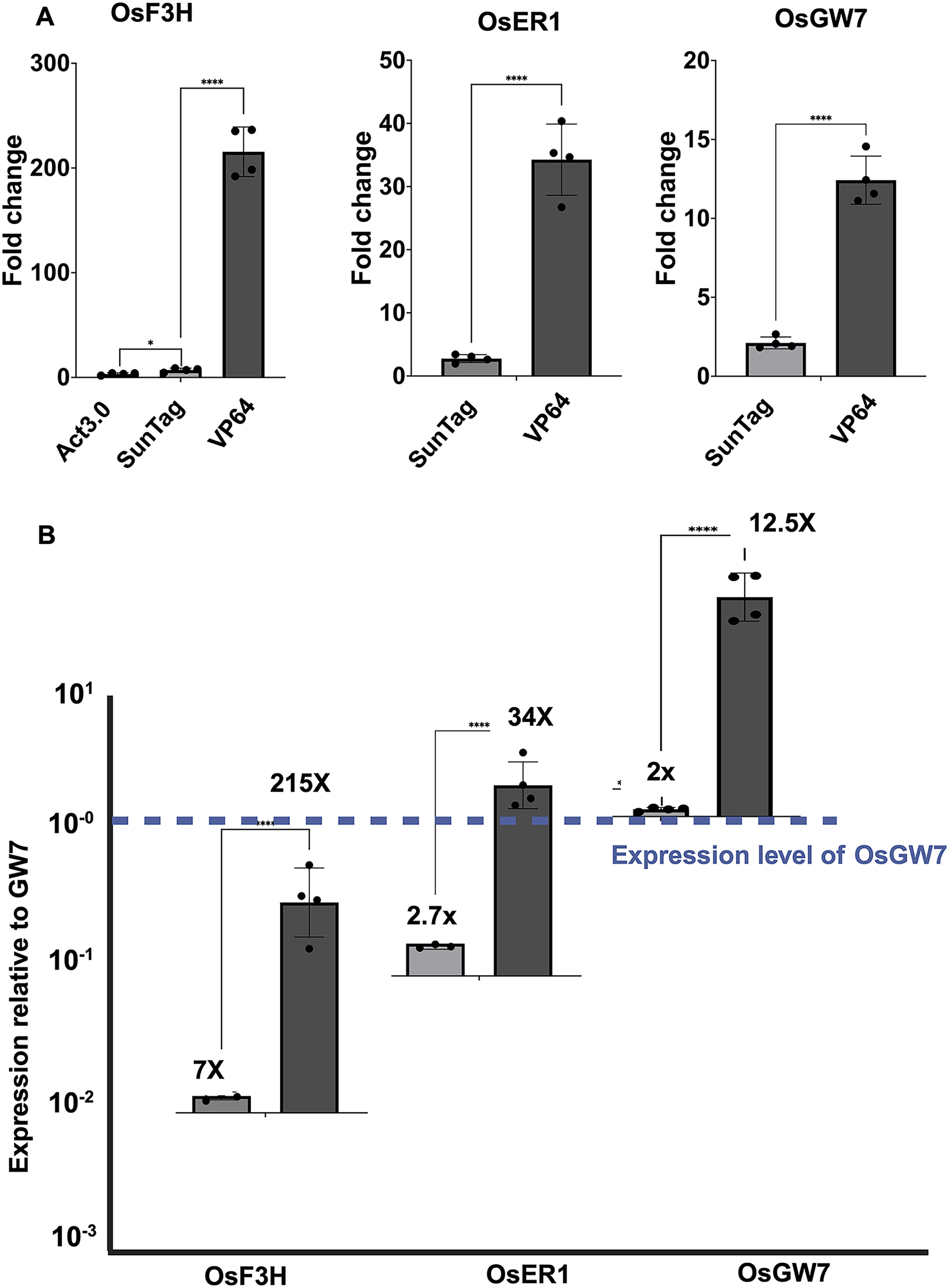
Comparison of CRISPRa effectors (VP64, SunTag, Act3.0) on the activation of rice target genes OsF3H, OsER1, and OsGW7.
Discussion
A versatile dual-luciferase assay to benchmark CRISPRa systems and scRNA designs
Our tetO-based dual-luciferase system offers a fast, quantitative, sensitive, and reproducible method to benchmark CRISPRa constructs and scRNA variants. This dual-cassette system can be reused in future studies to benchmark novel CRISPRa architectures against our new reference construct: dCas9 MCP-VP64, paired with the 5′ 2xMS2 scRNA and a composite promoter, is readily adaptable to other plant or nonplant systems. Although not tested here, the combination of a VP64 transcriptionally fused to a dCas9 with our system remains a potent alternative and may outperform our construct when maximal activation is required. 22 The use of alternative transcriptional activators, such as 2xTAD or TV, could further enhance the efficiency of CRISPRa.5,13
Boosting CRISPRa with a composite promoter
In most CRISPRa systems, scRNAs are expressed under a U3 promoter, driven by RNA polymerase III. However, low single-guide (sgRNA) expression has been shown to limit genome editing efficiency, particularly in suboptimal contexts. 24 In plants, composite promoters that combine Pol II and Pol III elements have been reported to significantly enhance prime editing efficiency.20,25,26 Here, we demonstrate that a composite promoter improves CRISPRa activity by an order of magnitude compared with a classical U3 promoter (see Fig. 2D, middle panel). Interestingly, previous studies using a Pol II promoter alone (e.g., pZmUBI in Act3.0) 5 failed to enhance CRISPRa, highlighting the importance of promoter architecture. The composite promoter used in our work combines Pol II (CaMV35S, CmYLCV) and Pol III (U6, tRNA) elements and flanks the scRNA with a 5' tRNA and a 3′ HDV ribozyme. 20 These features enable precise processing of the scRNA ends, ensuring proper folding and nuclear retention. In contrast, standard Pol II-driven expression often leads to 5' capping and 3' polyadenylation, which can mislocalize RNAs to the cytosol and may reduce functional efficacy in CRISPRa applications.
A novel, highly efficient tetraloop-2xMS2 scRNA
We developed a highly efficient scRNA containing two MS2 aptamers inserted in the tetraloop. This scaffold outperformed gR2.0, the reference scRNA of the SAM system. However, we initially expected that the scRNA with eight aptamers would be the most efficient. The SIRIUS strategy, originally developed to improve RNA imaging, stabilizes multimeric RNA scaffolds by introducing engineered stem loops that enhance the structural integrity of aptamer arrays. 23 This design enabled efficient recruitment of multiple GFPs and significantly improved fluorescent signal intensity. 23 Inspired by this, we adapted the approach to create novel scRNA scaffolds for CRISPRa. Despite our efforts, none of the aptamer configurations inserted at the 3′ end of the scRNA led to transcriptional activation (Fig. 2D, left panel). The failure of 3′-end aptamer insertion may be due to suboptimal recruitment or spatial positioning of the transcriptional machinery, possibly because the distal 3′ region of the scRNA is less accessible for effector complex formation, or due to interference with Pol II/Pol III processing. These findings suggest that SIRIUS-like stabilization alone is insufficient in eukaryotic CRISPRa, where aptamer position within the sgRNA architecture is critical for functional activation and is consistent with previous experiments describing inefficient or poorly efficient scRNA with aptamers inserted in 3′ end. 22
Structural considerations in scRNA design: scRNA dimerization of MCP-VP64 may explain differences in CRISPRa efficiency
Attempts to further enhance the 5′ 2xMS2 scaffold, either by inserting eight MS2 aptamers in the tetraloop or by adding two more in hairpin 2, did not improve activation. Since MCP-VP64 dimerization is key for CRISPRa, 27 we generated exploratory AlphaFold models of dCas9–scRNA–MCP-VP64 assemblies and computed mean Predicted Aligned Error (PAE) by domain (Supplementary Fig. S4A) from AlphaFold models via a custom script. We observed an inverse correlation between CRISPRa output and mean PAE for MCP-VP64 dimer regions (Supplementary Fig. S4). Given AlphaFold’s current limits on large RNA–protein assemblies and PAE values reflect model uncertainty, not experimental binding affinities, we interpret these in silico results as hypothesis-generating: one possibility is that high-copy MS2 arrays alter scRNA geometry in a way that disfavors MCP-VP64 dimer engagement as MCP-VP64 dimerization precedes RNA binding. 27 Although based on structural predictions, this approach offers a valuable in silico strategy to evaluate new scRNA designs for their ability to stabilize MCP-VP64 dimers. This suggests that highly efficient multimeric MS2 scRNAs could still be engineered, provided steric hindrance is minimized to preserve dimer interface integrity. This approach offers a useful prior to guide scRNA design, to be validated experimentally.
A streamlined CRISPRa design outperforms SunTag in rice
Several hypotheses can be proposed to explain why our simple MCP–VP64 system consistently outperforms SunTag-based CRISPRa constructs. First, our Act3.0 and SunTag constructs may not be fully functional due to differences in dCas9 sequence, sgRNA scaffold, or plasmid context compared with the original Act3.0 system. 5 However, since both constructs are able to activate endogenous genes, they are functional. Second, SunTag elements are notoriously challenging to express efficiently, possibly because of their repetitive nature, translational burden, or the instability/toxicity of scFv modules.16,28 In line with this, a recent study reported strong SunTag activation in protoplasts but markedly reduced activity in stable lines due to insufficient scFv accumulation, an observation that motivated the development of the MoonTag system. 16 Third, excessive recruitment of VP64 domains, whether through SunTag scaffolds or high-copy MS2 arrays, can interfere with CRISPRa activity by hindering dCas9 binding, inducing transcriptional squelching, or causing proteotoxic stress.27,29 In our hands, both SunTag and the 5′-8xMS2 scaffold underperformed relative to the streamlined 5′-2xMS2 configuration, consistent with the idea that overloading the activation complex can reduce transcriptional output.
In addition to these mechanistic considerations, experimental factors likely contribute to the apparent discrepancy between our results and those reported for Act3.0. The very strong inductions described in the original Act3.0 study 5 were obtained under exceptionally high transfection conditions (80 µg DNA in 150 µL protoplasts), corresponding to an estimated ∼20–30-fold higher DNA-per-cell ratio than in our standard conditions (10 µg in 500 µL). Such conditions greatly enhance transient expression of large, multicomponent architectures like Act3.0 and may artificially boost their reported performance. Consistent with this hypothesis, several independent studies using Act3.0 in other plant species report only modest activation for most endogenous genes, suggesting that Act3.0 requires unusually high expression levels to achieve strong induction.6,30,31 A parallel trend is observed in mammalian systems, where CRISPRa platforms based on RNA aptamers (e.g., SAM or MS2-recruiting activators) consistently outperform SunTag-based activators,32,33 underscoring the inherent efficiency limitations of scFv-dependent systems.
Scope and limitations
Our findings are derived from rice protoplast assays using plasmid-based expression. We did not quantify expression of individual modules nor scRNA abundance/localization. Accordingly, conclusions about stochiometric balance, promoter effects, and scRNA architecture should be viewed as condition-dependent and hypothesis-generating. Future work will extend validation stable in planta and at challenging, heterochromatic loci and will incorporate orthogonal CRISPRi modules. Given the generic nature of scRNA geometry and promoter architecture, the framework should be broadly portable across species. Finally, there are other transcriptional activators besides VP64 that appear to be more effective, such as the 6xTALE activation domain from Xanthomonas combined with two VP64 units, which outperforms earlier fusion constructs in both plant and mammalian cells.9,10
Tunable CRISPRa activation: Controlling gene expression across magnitudes
Our results thus suggest that a streamlined system, combining dCas9, MCP-VP64, an optimized sgRNA (5' 2xMS2), and a composite promoter, can induce robust CRISPRa of endogenous rice genes. Various combinations of scRNA and promoter (e.g., U3 or composite) can robustly activate gene expression over several orders of magnitude: from a 2-fold increase with U3/5′ 8xMS2 to up to a 100-fold activation with composite/5′ 2xMS2. Together, these results define a modular, scalable CRISPRa system that expands the toolbox for tunable gene regulation in plants, with direct applications in crop engineering and synthetic biology. Moreover, this system intended to be CRISPRi-compatible; future studies integrating orthogonal aptamer–effector pairs will be needed to establish CRISPRi performance within this framework. The ability to combine CRISPRa and CRISPRi within the same system opens possibilities in synthetic biology. Our synthetic promoter, combining the tetO sequence, a minimal p35 promoter, and a 5′ scRNA with 2×MS2, also provides an attractive option for designing programmable logic gates that enable precise, multilayered control of gene networks, 34 underscoring the potential of this streamlined design as a generic platform for both plant biotechnology and broader synthetic biology applications.
Authors’ Contributions
D.G., C.P., T.M., and A.-C.M. designed and built the CRISPRa and scRNA constructs. M.B. and D.G. performed the protoplast transformations, and together with C.P. carried out the dual-luciferase assays. C.C. performed RNA extractions and RT-qPCR. D.G. and C.P. drafted the article. All authors contributed to data analysis and reviewed and approved the final article.
Footnotes
Author Disclosure Statement
The authors have no relevant financial or nonfinancial interest to disclose.
Funding Information
This work was partially supported by the French National Agency of Research (ANR PRCI Greener ANR-20-CE20-0028) and the ARYZE project, supported by Methane Hub consortium.
Data Availability Statement
All data supporting the findings of this study are available within the article and its supplementary materials.
Ethics Approval Statement
All experiments were conducted in compliance with institutional, national, and international guidelines for the handling of genetically modified organisms and plant material.
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.