
Abstract
A major goal of clinically oriented CRISPR–Cas9-based applications is safe and effective in vivo gene editing (knockout or correction) with precise targeting. Substantial efforts have been devoted to the preclinical development of novel drug delivery platforms that enable efficient, targeted delivery. However, the immune responses induced by CRISPR–Cas9 treatment are often overlooked. Preexisting immunity to clinically relevant Cas9 proteins has already been established as a consequence of natural exposure to Cas9-bearing bacteria, which may implicate the safety and efficacy of CRISPR–Cas9-based therapies. Naturally, CRISPR–Cas9 therapies should be nonimmunogenic to avoid amplifying existing Cas9-specific immunity, especially cytotoxic T cell responses. Nonviral delivery systems, such as lipid nanoparticles (LNPs), are widely regarded as less immunogenic than more traditionally used viral vectors, even though LNPs are suitable as a vaccination platforms. In this study, we investigate the induction of SpCas9-directed immunity in C57BL/6 mice upon repeated dosing of LNPs encapsulating Cas9-coding mRNA in two different settings: (1) a vaccination-resembling setting using intramuscularly administered adjuvanted LNPs, and (2) a therapy-resembling setting using intravenously injected, liver-targeting LNPs. In both settings, Cas9-specific T cell responses were detected by evaluating increased total IFN-γ levels upon ex vivo restimulation of isolated splenocytes. However, undetectable Cas9-reactive antibodies induced in the therapeutic setting emphasize the discrepancy between humoral and cellular responses. To improve future monitoring of Cas9-specific T cell responses, we report six Cas9-derived epitopes recognized by CD8+ T cells, as well as a CD4+ T cell polypeptide carrying one of the CD8+ T cell epitopes that induced strong IFN-γ production ex vivo. This work is intended to facilitate the preclinical monitoring of Cas9-specific T cell responses in C57BL/6 mice and support the development of safe CRISPR–Cas9-based therapies.
Introduction
Genome editing in vivo using CRISPR–Cas9 and next-generation CRISPR technologies, such as base editing and prime editing, holds great promise for treating and even curing debilitating genetic diseases. By delivering the therapeutic components—a Cas9 nuclease (or alternative) and a guide RNA—directly into the target cell, they can correct disease-causing mutations. However, the immunogenicity of these components, especially the Cas9 protein, poses a challenge in the translation of such therapies into clinical use.1,2 Therapeutically relevant Cas9 variants originate from bacteria such as Streptococcus pyogenes and Staphylococcus aureus.3–5 Humans are naturally exposed to these Cas9-bearing bacteria, and consequently exhibit preexisting SpCas9- and SaCas9-specific immune responses, including anti-Cas9 serum antibodies6–9 and Cas9-specific CD4+ and CD8+ T cells.6,10,11 The existence of Cas9-reactive CD8+ (cytotoxic) T cells is particularly concerning for CRISPR–Cas9 therapies due to the intracellular presence of the Cas9 protein. Cytosolic proteins, including Cas9, are continuously degraded by proteases into peptides. 12 These peptides can be loaded onto major histocompatibility complex (MHC) class I molecules and expressed on the cell surface for recognition by cytotoxic T cells. Epitope recognition results in cytotoxic T cell activation and proliferation, ultimately leading to inflammation and T cell-mediated target cell killing, resulting in the loss of therapeutic efficacy.11,13 Short-term administration of immunosuppressive drugs such as dexamethasone and histamine blockers could be used to minimize immune activation; however, this may not be sufficient to prevent Cas9-directed immune responses and associated inflammation. 13
Preclinical studies have reported successful CRISPR–Cas9-based gene editing of different target cells and tissues using LNPs, including hepatocytes in the liver (intravenous [IV] injection),14–18 skeletal muscle cells (intramuscular [IM] injection), 19 retinal pigment epithelial cells (intravitreal injection), 20 and multiple cell types in the lungs. 21 Laboratory mice are commonly used in preclinical studies to investigate the treatment benefits before proceeding toward clinical translation. However, they are raised in specific-pathogen-free conditions and are therefore minimally exposed to SpCas9. Administration of Cas9 therapies in naive mouse models therefore represents de novo priming of immune responses, whereas in humans with preexisting immunity, Cas9 administration could reactivate existing Cas9-specific T cells. Whether preexisting SpCas9-specific immune responses in humans hamper the beneficial effects of gene therapy remains undetermined. Furthermore, it remains unclear whether repeated dosing of gene therapies, which may be crucial for treatment safety and efficacy, 22 would further amplify the immune responses. A preclinical study using mice preimmunized with SaCas9 demonstrated an increase in cytotoxic T cells in SaCas9-immunized mice receiving liver-targeting AAV–CRISPR therapy. 23 However, the study did not directly assess whether T cell induction was Cas9- or vector-specific. In canine models, AAV-mediated Cas9 expression induced Cas9-specific immunity and inflammation, which was Cas9-specific despite immune suppression. 13 These results emphasize the importance of investigating Cas9-specific immune effects in in vivo gene therapies and the occurrence of Cas9-specific T cells.
A multitude of factors can contribute to the induction or amplification of Cas9-directed immune responses in gene therapy in vivo, including the choice of CRISPR–Cas9 delivery vector, 19 administration route, target cell types or tissues, and disease state. 13 LNPs have been proposed as a less immunogenic alternative14,19,24 to adenoviral vectors.25–28 Nevertheless, several studies have found that both empty and mRNA-encapsulating LNPs can induce immune activation.29–32 While this is beneficial for immunization against infectious diseases such as COVID-19, immune activation during genome editing is undesired. In this study, we investigated the induction of Cas9-specific T cells and antibodies in C57BL/6 mice repeatedly injected with LNPs composed of clinically approved ionizable lipids and encapsulating SpCas9-coding mRNA. Furthermore, we predicted and experimentally evaluated SpCas9-derived epitopes specific for MHC class I (H-2Kb/d) and class II (I-Ab) for recognition by CD4+ or CD8+ T cells in C57BL/6 mice.
De novo priming of SpCas9-directed immune responses was evaluated in healthy mice undergoing repeated LNP dosing under two scenarios: (1) IM immunization with adjuvanted LNPs to mimic a vaccination regimen and (2) systemic administration of nonadjuvanted LNPs primarily targeting the liver to resemble a gene therapy setting. In the immunization scenario, poly(I:C)-adjuvanted LNPs (containing ALC-0513) were injected IM thrice to induce Cas9-directed T cell responses required to identify Cas9-specific epitopes recognized by CD4+ and CD8+ T cells in this mouse strain. On the other hand, DLin-MC3-DMA-based liver-targeting LNPs were injected IV thrice, closely representing hepatic gene therapy. Upon restimulation of splenocytes, SpCas9-specific T cell responses were detected in both experimental groups. In contrast, SpCas9-specific antibodies were nondetectable or low in mice that received liver-targeting LNPs, suggesting a discrepancy between humoral and cellular immunity. Moreover, through prediction and experimental evaluation of SpCas9-derived MHC I and MHC II-binding peptides, we identified six H-2Kb/d peptides that induced activation of at least 2% of cytotoxic T cells, and one MHC class II (I-Ab)-binding polypeptide sequence that induces strong IFN-γ release upon splenocyte restimulation. These findings provide insight into LNP-induced immune responses in preclinical models and monitoring of SpCas9-recognizing T cells, especially the induction of Cas9-directed cytotoxicity.
Materials and Methods
SpCas9-derived epitopes with MHC class I-binding sequences
Prediction of MHC class I-binding SpCas9-derived peptides was performed using the NetMHCpan 4.1 tool. 33 The prediction was performed for the MHC class I alleles H-2-Db and H-2-Kb, haplotypes carried by C57BL/6 mice, and included multiple peptide lengths ranging from 8 to 14 amino acids and different binding affinities. Retrieved peptides, sorted based on a percentile rank EL < 2.000, yielded 143 H-2-Db and 132 H-2-Kb peptides. On top of that, an affinity threshold of 500 nM further reduced the number of predicted peptides to 14 H-2Db- and 51 H-2Kb-binding peptides. Overlapping peptides with highly similar cores were inspected, and the one with the highest binding affinity was selected. Lastly, 22 peptides (4 H-2Db- and 18 H-2Kb-binding peptides) (Supplementary Table S1) were synthesized by SynPeptide Co., Ltd., China, at >95% purity.
SpCas9-derived epitopes with MHC class II-binding sequences
Prediction of MHC class II-binding Cas9-derived peptides was performed using the epitope prediction tool accessible through the Immune Epitope Database (IEDB). 34 Murine H2-Ab1-binding peptides with a predicted percentile rank of ≤ 6 were considered potential epitopes, resulting in the identification of 45 peptides distributed across nine domains of the SpCas9 protein sequence. We selected all nine domains, referred to as polypeptides, for CD4+ T cell stimulation. Before polypeptide synthesis, flanking residues were added to the core of each domain by extending the amino acid sequence at the N- and/or C-terminus of the predicted peptide (Supplementary Table S2). The final polypeptides were synthesized by SynPeptide Co., Ltd., China, at >95% purity.
Design of RNA constructs for enhanced antigen presentation
For the induction of antigen-specific immune responses, antigen presentation is crucial. 35 The trafficking of de novo synthesized antigen from the cytoplasm toward endosomal compartments associated with MHC processing is enhanced by the inclusion of protein sequences derived from proteins that naturally colocalize with antigen presentation compartments.35,36 We designed a construct containing a signaling peptide (5ʹ end) and cytosolic and transmembrane regions (3ʹ end), flanking the target antigen and fused by the inclusion of GS linkers (Supplementary Fig. S1A). In this study, two target antigens were used: (1) a protein sequence derived from chicken ovalbumin (OVA) protein (OVA139–386) was used as a model antigen to assess antigen presentation 36 and (2) human codon-optimized full-length SpCas9 was used to induce Cas9-specific immune responses in mice.
The pCMV6-XL5 vector was constructed by inserting the T7 promoter and the 5′ UTR and 3′ UTR regions flanking the multiple cloning site, as reported before. 37 Consequently, the target antigens OVA139–386 or SpCas9 containing antigen processing sequences were cloned into the open reading frame, flanked by the 5′ and 3′ UTR regions.
First, the OVA139–386-containing construct was generated. The N-terminus of OVA139–386 was fused to the signaling peptide sequence of the invariant chain (GenBank APD15604.1, 1–80) as reported before. 38 To improve the insertion of newly synthesized protein into the membrane of the endosomal trafficking compartment, the C-terminus of OVA139–386 was fused to the transmembrane (TM) and cytosolic (CS) domains of CD27, referred to as TM/CS (GenBank M63928, 565–651 nt). 35 GS-based linkers (SGGGGS/agtggcggcggaggatcc) were inserted between the sequences to improve protein folding. 39 The final construct (Ii-OVA-TM/CS) contained a start codon (ATG) at the 5ʹ site and two stop codons (TAG TAA) at the 3ʹ site. The newly designed DNA construct was ordered as gBlock (IDT) and cloned into the polymerase chain reaction (PCR)-amplified backbone of the pCMV6-XL5-T7 plasmid between the UTR regions by InFusion cloning (Takara Bio, Europe). The obtained plasmid pCMV6-XL5-T7-(Ii-OVA-TM/CS) was used as a template for replacing the OVA139–386 sequence with the SpCas9 sequence. Briefly, the gene for Cas9 (Csn1) endonuclease from the Streptococcus was PCR-amplified from lentiCas9-Blast (Addgene plasmid #52962, 4–4104 nt) and inserted in between the GS linkers by InFusion cloning (Takara Bio). The final constructs were confirmed by sequencing.
When antigen presentation was not the primary interest, a commonly used wild-type (WT) SpCas9 sequence containing a nuclear localization signal (NLS), without the mentioned molecular modifications, was used as described before 15 and will be referred to as SpCas9-NLS.
RNA synthesis by in vitro transcription
For Ii-OVA-TM/CS and Ii-SpCas9-TM/CS mRNA, the newly constructed plasmids were linearized by SalI and extracted from the gel using a PureLink Quick Gel Extraction Kit (Invitrogen, Thermo Fisher Scientific, USA). The linear DNA templates were used for gene polyadenylation using Tail PCR primers, of which the reverse primer contains TTTTTT at the CTTC end. Tail PCR was performed using Q5 DNA polymerase. The reaction was then applied to a PCR program: 98°C 3 min, (98°C for 30 s, 69°C for 15 s, and 72°C for 30 s (OVA) or 80 s (SpCas9) for 30 cycles, followed by a final extension at 72°C 10 min. The PCR mixture was purified with a PureLink PCR Purification Kit (Invitrogen, Thermo Fisher Scientific) to obtain DNA templates for in vitro RNA (IVT) synthesis. SpCas9 mRNA used for liver-targeting LNPs was prepared as described before. 15
mRNA was synthesized using the VENI All-in-One mRNA Synthesis Kit with Cap1 Analog (Leish Bio, Utrecht, The Netherlands). Briefly, 2 μg of purified tail PCR product was provided as a template. The final nucleotide concentrations in the reaction were 6 mM for the Cap1 analog and 7.5 mM for adenosine triphosphate, guanosine triphosphate, cytidine triphosphate, and N1-methylpseudouridine triphosphate. The reaction mixture was treated with Turbo DNase and purified using the Monarch Spin RNA Cleanup Kit (500 μg) (NEB). mRNA was stored at −20°C for the short term until further use.
Lipid nanoparticle formulation
Ionizable lipid (either DLin-MC3-DMA or ALC-0315), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG) were separately dissolved in ethanol and then combined at 50:38.5:10:1.5 molar ratio, respectively, at a total lipid concentration of 2–8 mM (0.3–1.2 mg/mL) in 90% ethanol/10% citrate buffer (25 mM, pH4). RNA molecules were dissolved in citrate buffer (25 mM, pH 4). Briefly, Ii-OVA-CS/TM, Ii-SpCas9-CS/TM, and SpCas9 mRNA molecules were produced as described. CCR5 sgRNA (TCAGTCTATACCCGATCCAC, molecular weight [MW] = 32249.7 g/mol) (IDT, Europe) was dissolved in nuclease-free water at 20 μM.
For optimization of adjuvanted ALC-0315-LNPs, Ii-OVA-TM/CS mRNA (435 kDa) alone, poly(I:C) alone, or both components together at mass ratios of 500:1, 100:1, 20:1, or 10:1 of Ii-OVA-TM/CS mRNA to poly(I:C) were prepared in the aqueous solution. For ALC-0315 LNPs used in animal experiments, Ii-OVA-TM/CS mRNA and Ii-Cas9-TM/CS mRNA (1522 kDa) were combined for encapsulation in the same LNP at either a 1:1 (n/n) or 1:3.5 (w/w) ratio. Synthetic double-stranded (ds)RNA analog poly(I:C) (HMW, 1.5–8 kb) was included at a 500:1 (w/w) ratio of total RNA (Ii-OVA-TM/CS + Ii-Cas9-TM/CS) to poly(I:C). For DLin-MC3-DMA LNPs used in animal experiments, SpCas9-NLS mRNA (1436 kDa) and sgRNA (32 kDa) were combined at either a 1:23.6 (n/n) or 2:1 (w/w) ratio. All RNA molecules were dissolved in citrate buffer (25 mM, pH 4) at a concentration of 25–64 μg/mL. The N/P ratio between the ionizable lipid and RNA was 7:1 or 6:1 for ALC-0315-LNPs or Dlin-MC3-DMA-LNPs, respectively. In case of ALC-0315-LNPs encapsulating only poly(I:C), the same amount of poly(I:C) was included as in the formulation containing a 20:1 OVA mRNA:poly(I:C).
LNPs were prepared by rapid mixing of the aqueous RNA solution and ethanolic lipid solution using microfluidic mixing at a volume ratio of 1:3 and a total flow rate of 10,000 μL/min. Pre-LNPs were dispensed at a volume 0.8–1.5 mL and immediately diluted six times in 1× phosphate-buffered saline (PBS). The diluted LNP solution was loaded onto prewashed Vivaspin® 6 Centrifugal Concentrator PES columns (10 kDA MWCO) (Sartorius, UK), centrifuged at 3000 × g at room temperature (RT) for 15–30 min, and resuspended in fresh PBS. The washing step was repeated until the ethanol content was diluted 250-fold. Purified LNPs at an RNA concentration of 20–100 ng/μL were stored at 4°C until further use.
Physical characterization of LNPs
The average diameter (nm) and PDI of LNPs were determined by dynamic light scattering using a Zetasizer Nano S (Malvern ALV CGS-3, Malvern, UK) using a semi-microcuvette (Sarstedt Inc., Germany). Measurements were performed at a scattering angle of 173°, with the measurement position set to 4.65 mm. LNP samples were prepared by diluting the LNPs in PBS (10–60× dilution), calibrating the samples at RT for 5 min before measurement.
RNA encapsulation assay (RiboGreen)
RNA encapsulation efficiency was determined using the Quant-it™ RiboGreen Reagent and RNA Assay Kit (R11491, Thermo Fisher Scientific) according to the manufacturer’s recommendation. Briefly, TE buffer was prepared with or without 1% Triton X-100 in nuclease-free water. LNPs were dissolved in 50 μL of TE buffer with or without Triton X-100, vortexed, transferred to a 96-well plate, and incubated for 15 min before adding TE buffer to a final volume of 100 μL. RNA standard curves, ranging in final RNA concentration between 0 and 200 ng/μL RNA were prepared in 100 μL either without Triton X-100 or with 0.5% Triton X-100 for a final Triton X-100 concentration of 0.25%. A working solution was prepared by diluting the Quant-iT RiboGreen RNA Reagent in TE buffer at a 1:2000 dilution. One hundred microliters of the working solution was added to each well containing either a standard or a sample, followed by incubation on a shaker protected from light for 10 min. Fluorescence was measured using a fluorescence plate reader FP-8300 Spectrofluorometer (Jasco, UK) at 480 nm/520 nm excitation/emission wavelengths, respectively.
Gel retardation assay
A gel retardation assay was performed to examine the release of (non)encapsulated RNA from the LNPs and to confirm the results obtained from the RiboGreen assay. LNPs were either disrupted with Triton X-100 or left untreated. For LNP disruption, LNPs were resuspended in PBS containing 1% Triton X-100 and heated at 80°C for 10 min before being transferred on ice. Treated and untreated LNPs were mixed with 1× loading dye, loaded onto a 1% agarose gel stained with Midori Green Advance DNA/RNA Stain (Nippon Genetics Europe), and run at 100 V for 30 min in 1× TAE buffer (pH 8) (Bio-Rad Laboratories B.V., Veenendaal, The Netherlands). The gel was imaged using the ChemiDoc Imaging System (Bio-Rad Laboratories B.V.).
Cell culture
Bone marrow cells were collected from murine (C57BL/6 WT) femurs and tibias by flushing with complete Iscove's Modified Dulbecco’s Medium (IMDM) (Gibco, Thermo Fisher Scientific, Landsmeer, The Netherlands) supplemented with 10% fetal calf serum (FCS) (Bodinco, Alkmaar, The Netherlands), 100 units/mL of penicillin (Gibco, Thermo Fisher Scientific), 100 μg/mL of streptomycin (Gibco, Thermo Fisher Scientific), and 0.5 µM β-mercaptoethanol (Gibco, Thermo Fisher Scientific) using a 21 G needle. Bone marrow cells were seeded in 6-well plates at a cell density of 450,000 cells/mL in complete IMDM. Cells were cultured at 37°C and 5% CO2 in the presence of granulocyte-macrophage colony-stimulating factor (in-house produced) for 7 days. Differentiated bone marrow-derived dendritic cells (BMDCs) were collected using a cell scraper and seeded for further experiments.
The OVA257–264-specific, H-2Kb-restricted CTL hybridoma B3Z is a cell line developed by Karttunen et al. 40 B3Z cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 units/mL of penicillin (Gibco, Thermo Fisher Scientific), 100 μg/mL of streptomycin (Gibco, Thermo Fisher Scientific), and 0.5 µM β-mercaptoethanol (Gibco, Thermo Fisher Scientific). All cells were cultured in a humidified atmosphere (5% CO2, 37°C).
BMDCs stimulation
To detect BMDC stimulation, 150,000 BMDCs were seeded into a F-bottom 96-well plate in 100 μL of cIMDM and incubated at 5% CO2, 37°C for 2 h to allow attachment to the plate. LNPs were resuspended in complete IMDM and added to the cells to a final RNA concentration of 100 ng per well. After 16 h, the plate was centrifuged at 350 × g for 5 min at 4°C; the medium was removed, and the cells were washed with 1× PBS and centrifuged again at 350 × g for 5 min at 4°C. Cells were blocked for 10 min with 10 μg/mL Fc Block (2.4G2, in-house produced). BMDCs were stained with CD11c-FITC (1:200, eBioscience, #11-0114), CD86-PE-Cy5 (1:800, eBioscience, #15-0862), MHC II-eFluor (1:400, eBioscience, #485321). and ViaKrome808 (1:1000, Beckman Coulter, Indianapolis, IN, USA) in staining buffer (1× PBS supplemented with 2% FCS and 0.01% sodium azide). After 30 min incubation at 4°C in the dark, cells were washed with PBS three times and resuspended in 100 µL of staining buffer for measurement. Relevant single-stain and fluorescence-minus-one controls were included. Samples were recorded on a Beckman Coulter CytoFLEX LX, and data were analyzed using FlowJo Software v.10.7 (FlowJo LLC, Ashland, OR, USA).
In vitro Ag cross-presentation by BMDCs
To assess antigen presentation by BMDCs and T cell recognition, BMDC-B3Z T cell co-cultures were performed as described.40,41 Briefly, 40,000 BMDCs were seeded into an F-bottom 96-well plate and left to adhere for 2 h. LNPs were resuspended in fresh complete IMDM and added to the cells to a final RNA concentration of 100 ng per well. After 4 h, 100,000 B3Z T cells were added per well and incubated overnight for 16–20 h. After incubation, the plate was centrifuged at 350 × g for 5 min at RT. The medium was removed, and the cells were washed with 1× PBS and centrifuged at 350 × g for 5 min at RT. The supernatant was then removed, and 100 µL of 0.15 mM CPRG solution in CPRG lysis buffer (0.5% NP-40 in 1× PBS) was added. The plate was incubated at RT in the dark for 7–16 h. The absorption was measured at 570 nm, with 650 nm as the reference wavelength, using a microplate reader.
Mice
Mice used for bone marrow isolation (8-week-old C57BL/6 mice WT, male or female) and mice used in the animal experiment to study SpCas9-specific immune responses (8-week-old C57BL/6 mice WT, female) were purchased from Charles River Laboratories (Germany). Mice were housed at the Central Laboratory Animal Research Facility of Utrecht University. Upon arrival at the facility, mice were acclimatized for 1 week. Animals were kept under standard conditions of the animal facility. Food and water were provided ad libitum. All experiments were approved by the Animal Experiment Committee of Utrecht University (AVD10800202115687).
Induction of SpCas9-specific immune responses in SPF mice
Mice were randomized into three groups based on weight. Mice received either IV administration (200 μL) of PBS, IM administration (50 μL) of ALC-0315-based LNPs encapsulating OVA and SpCas9 mRNA (1:1, w/w), and poly(I:C) (1:500, n/n, total RNA:poly(I:C)) (n = 6), or IV administration (200 μL) of Dlin-MC3-DMA-based LNPs encapsulating SpCas9 mRNA:sgRNA (2:1, w/w; n = 6). All mice were injected on days 0, 7, and 14. Blood was drawn via cheek puncture on the same days (0, 7, and 14) before the injections. The experiment was terminated on day 20, and spleens, iliac lymph nodes, and blood were collected. Organs were stored in ice-cold PBS for <2 h until further processing. Blood samples were collected in 0.8 mL z-serum separation tubes (Greiner Bio-One, Kremsmünster, Austria) and allowed to clot at RT for 30 min before being transferred to 4°C for <2 h. Serum was separated from blood cells by centrifugation at 10,000 × g for 10 min at 4°C. Serum was collected into 1.5 mL Eppendorf tubes and stored at −20°C until measurement.
Single-cell suspension from spleen and lymph nodes
Iliac lymph nodes from mice within the same group were pooled before processing. Spleen or pooled lymph nodes were mechanically processed by mashing through a 70-micron cell strainer (Corning, New York, USA) using a syringe plunger and washed with 5 mL of ice-cold RPMI medium (Gibco, ThermoFisher Scientific). The resulting single-cell suspension was centrifuged at 300 × g for 5 min at 4°C, and the supernatant was discarded. Splenocytes were resuspended in 1 mL of ice-cold ammonium-chloride–potassium lysis buffer to lyse the erythrocytes. After 2 min, 9 mL of ice-cold medium was immediately added to the cell-lysis buffer suspension and centrifuged at 300 × g for 5 min at 4°C. The pellet was resuspended in fresh medium.
Ex vivo restimulation with MHC class I-binding peptides
Splenocytes were seeded at a density of 500,000 splenocytes per well in a 96-well U-bottom plate (Falcon) in 150 μL cRPMI. Fifty μL of medium containing either 1 μg/mL OVA-derived peptide SIINFEKL (vac-sin, InVivoGen), 20 ng/mL PMA and 1 μg/mL ionomycin (positive control), or plain medium was added to a final volume of 200 μL. To identify SpCas9-derived MHC class I-binding peptides, 200,000 pooled lymph node cells were seeded per well into 96-well U-bottom plate (Falcon) and stimulated either with 20 ng/mL PMA and 1 μg/mL Ionomycin (positive control), 1 μg/mL of each peptide (Cas9-derived MHC class I peptide or SIINFEKL as irrelevant peptide control), or left unstimulated, in a total volume of 200 μL. After 2 h of incubation at 37°C and 5% CO2, 40 μL Brefeldin A was added to the cells to a final concentration of 4 ng/μL to block the secretion of cytokines from the cells. The plate was incubated for an additional 4 h at 37°C and 5% CO2. After the incubation, the cells were stained for flow cytometry as described below.
Ex vivo restimulation with proteins or MHC class II-binding polypeptides
Splenocytes were seeded at a density of 500,000 splenocytes per well and stimulated either with 10 μg/mL Concanavalin A (positive control), 10 μg/mL SpCas9, 10 μg/mL OVA, 10 μg/mL of each SpCas9-derived MHC class II peptide, or left unstimulated. The plate was incubated at 37°C and 5% CO2 for 3 days. The supernatants were frozen at −80°C for cytokine analysis using Enzyme-Linked Immunosorbent Assay (ELISA).
Flow cytometry staining
After restimulation, cells were centrifuged at 300 × g for 5 min at 4°C. The cells were washed once with PBS and stained with the viability dye Viakrome 808 (1:1000, Beckman Coulter) in PBS. Cells were washed again with PBS, resuspended in 25 μL of Fc-blocking antibodies, and blocked for 10 min on ice. The surface staining was prepared by diluting fluorescently labelled antibodies in staining buffer (1× Dulbecco’s Phosphate Buffered Saline, DPBS [Gibco, ThermoFisher Scientific] supplemented with 2% FCS and 0.005% sodium azide). Each sample was stained with 50 μL of staining buffer containing CD4-eFluor 450 (1:400, clone RM4-5, eBioscience), CD8a-FITC (1:200, 16-10A1, BD Biosciences), CD25-BV650 (1:400, PC61, Biolegend), and CD3ε-APC (1:200, 145-2C11, BD Biosciences) for 30 min at 4°C. Subsequently, cells were washed twice with PBS, and intracellular staining was performed using the eBioscience™ Foxp3/Transcription Factor Staining Buffer Set according to the manufacturer’s instructions. Briefly, the cells were fixed using fixation/permeabilization solution for 30 min at room temperature, protected from light, and then centrifuged at 450 × g for 5 min at RT. Cells were washed twice with 1× permeabilization buffer. Intracellular staining solution containing IFN-γ-PE (1:300, XMG1.2, BD Biosciences) was prepared in 1× permeabilization buffer, and 50 μL was added to the cells, after which they were incubated for 30 min at 4°C in the dark and then washed twice with 1× permeabilization buffer. The cells were resuspended in 100 μL of staining buffer and recorded on a Cytoflex LX flow cytometer (Beckman Coulter). Data were analyzed using FlowJo Software v.10 (FlowJo Treestar, LLC, Ashland, OR, USA).
IFN-γ detection in the supernatants by ELISA
ELISA was performed to detect the IFN-γ released into the supernatants to indirectly assess T cell activation after 3-day restimulation. Briefly, F-bottom Costar 96-well plates (Corning, Kennebunk, ME, USA) were coated with a capture antibody anti-IFN-γ (1:250, BD Biosciences, cat. no. 551309) at 4°C overnight. The plate was washed three times with 0.01% Tween-20 in PBS and blocked with 1% Bovine Serum Albumin (BSA) in 1× DPBS for 30 min at RT while shaking. The supernatants frozen at −80°C were thawed at room temperature. The plates were washed, and 100 μL of nondiluted or 4× diluted supernatant or standard cytokine IFN-γ (PMC4031, Gibco) was incubated for 1 h at RT. The plates were then washed, and biotinylated detection antibody anti-IFN-γ (1:250, BD Biosciences, cat. no. 554410) together with streptavidin-HRP (BD Biosciences) were added. Plates were incubated for 1 h at RT while shaking. The plate was washed, and 50 μL of TMB substrate solution (BioLegend, 421101) was added per well. The reaction was stopped with 25 μL 2 N H2SO4 solution, and the OD was measured at 450 and 595 nm using an iMark™ Microplate Absorbance Reader (Bio-Rad).
Anti-Cas9 antibody detection in the mouse sera by ELISA
ELISA was performed to detect anti-Cas9 antibodies in mouse sera collected on days 0 (before injections), 14, and 30. Briefly, F-bottom Costar assay 96-well plates (Corning, Kennebunk, ME, USA) were coated with 1 µg/well SpCas9 (in-house produced) at 4°C overnight. The next day, the plates were washed three times with 0.01% Tween-20 in PBS and blocked with 100 µL of 1% BSA for 2 h at RT. All serum samples were diluted to 1:2000 in 1% BSA. A standard curve was prepared by diluting the anti-Cas9 monoclonal IgG1 antibody (clone 7A9; A-9000, Sanbio) in a four-step serial dilution (103–6.6 × 107). One hundred μL of either diluted serum sample (triplicates) or standard antibody dilution (in duplicates) was added per well, and the plates were incubated for 5 h at 4°C while shaking at 0.5xg. Plates were then washed, and 100 μL of HRP-labeled goat anti-mouse IgG1 (554002, BD Biosciences), diluted 1:1000 in 1% BSA, was added to each well. The plates were incubated at RT for 1 h while shaking. The plate was washed, and 50 μL of TMB substrate solution (421101, BioLegend) was added per well. The reaction was stopped with 25 μL 2 N H2SO4 solution, and the OD was measured at 450 and 595 nm using an iMark Microplate Absorbance Reader (Bio-Rad).
ELISA analysis
The average background optical density (OD) was subtracted from each triplicate reading. The mean and standard deviation were calculated for each mouse at each time point. To confirm the presence of SpCas9-specific antibodies, a threshold of three standard deviations above the mean of day 0 was established as the cutoff value. For data visualization, background-subtracted OD values from days 14 and 30 were normalized to OD on day 0 for each mouse (normalized value = 1).
Results
Optimization of poly(I:C)-to-RNA ratio in LNPs for antigen presentation and co-stimulation
For in vivo antigen delivery using LNPs, we constructed and synthesized nucleoside-modified mRNA molecules encoding either the full-length spCas9 protein or a partial sequence of the model antigen OVA, OVA139–386, which contains both CD8+ and CD4+ T cell epitopes (Supplementary Fig. S1A). To induce the strong antigen-specific immune responses required for epitope identification, the dsRNA analog poly(I:C), a TLR3 agonist, was chosen for co-encapsulation with mRNA into LNPs. To determine the optimal ratio of RNA-to-poly(I:C), we formulated LNPs containing both cargoes at different (weight) ratios. OVA139–386 RNA was encapsulated into LNPs either alone or in the presence of poly(I:C) at weight ratios of 10:1, 20:1, 100:1, and 500:1 (Supplementary Fig. S1B). The LNPs exhibited comparable physicochemical properties in terms of size, polydispersity, and encapsulation efficiency (Supplementary Fig. S1C). BMDCs, differentiated from bone marrow cells of C57BL/6 mice, were used for LNP transfection. To assess whether CD8+ T cells recognized the delivered antigen, BMDCs were co-cultured with B3Z T cells, which recognize the OVA-derived epitope SIINFEKL when presented on MHC class I by BMDCs. Antigen–MHC recognition by the T cell receptor, coupled to β-galactosidase, was measured by colorimetric conversion of CPRG. While LNPs without poly(I:C) enabled T cell recognition, poly(I:C) diminished this recognition in a concentration-dependent manner, even with equal amounts of RNA (Supplementary Fig. S1D). Even at the lowest poly(I:C) concentrations, we detected increased CD86 marker expression, indicating co-stimulation (Supplementary Fig. S1E). LNPs containing an mRNA to poly(I:C) ratio of 500:1 (w/w) were chosen for antigen-specific T cell induction in vivo due to the optimal balance between antigen recognition and immunostimulation.
Detection of SpCas9-directed immune responses using adjuvanted LNPs
For successful identification of antigen-derived peptides, a strong adaptive immune response to a target antigen is crucial. For this purpose, we used adjuvanted LNPs encapsulating antigen-coding mRNA to induce immune responses against the SpCas9 protein and the model antigen OVA139–386 in C57BL/6 mice. We formulated ALC-0315 lipid-based LNPs encapsulating SpCas9/OVA mRNA (1:1 molar ratio) and poly(I:C) at a ratio of 500:1 (Fig. 1A). The LNPs exhibited a diameter of around 95 nm, low polydispersity (0.11), and an encapsulation efficiency of around 95% (Fig. 1B), which was confirmed by visualizing the RNA using a gel retardation assay (Supplementary Fig. S2). Mice received IM immunizations of the LNPs three times (2 μg RNA per administration), given 1 week apart (Fig. 1C). Six days after the last immunization, splenocytes were isolated and restimulated ex vivo in the presence of spCas9 or OVA protein, or unstimulated, for 3 days. Antigen-specific T cell activation was measured as the concentration of IFN-γ secreted into the extracellular environment. Elevated IFN-γ levels were detected in the supernatants from all six mice following restimulation with either SpCas9 or OVA protein, whereas the untreated conditions exhibited background values (Fig. 1D). To assess the antigen-specific CD8+ T cell responses, splenocytes were restimulated ex vivo for 6 h with OVA peptide or left unstimulated. A significantly higher frequency of IFN-γ+ cells was detected among CD8+ CD3e+ T cells stimulated with pOVA as compared to the control (Fig. 1E), proving successful induction of antigen-specific cytotoxic T cells. Importantly, upon restimulation of splenocytes from PBS-injected mice with OVA or SpCas9 proteins, we did not detect above-background IFN-γ concentration (data not shown).
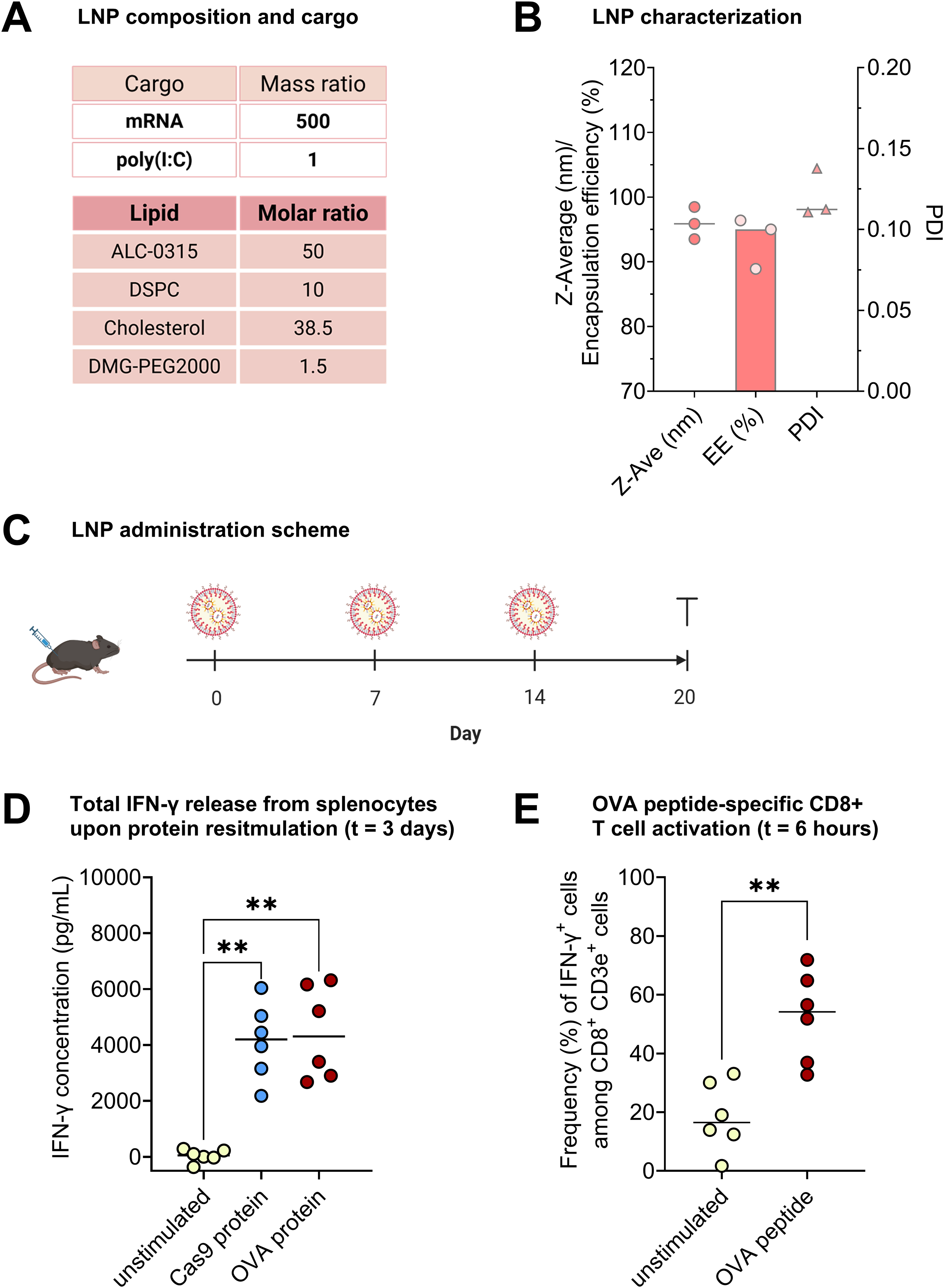
Induction of SpCas9-directed immune responses with adjuvanted LNPs in C57BL/6 mice.
SpCas9-derived MHC class I- and II-binding peptide prediction and selection
CD8+ T cells recognize MHC class I-presented epitopes derived from intracellular proteins, including SpCas9 protein, 42 and can initiate cytotoxic immune responses and antigen-specific cell clearance. MHC class I-binding SpCas9-specific peptides, 8–13 amino acids in length, were predicted using the NetMHCpan 4.1 server, 33 and 22 peptides conforming to the C57BL/6 mouse MHC class I-binding motif were used for further experimental testing (Supplementary Table S1).
CD4+ T cells orchestrate adaptive immune responses by recognizing epitopes presented on MHC class II molecules displayed on the surface of APCs. Even though MHC class II-presented peptides are traditionally derived from extracellular proteins, APCs can also present the epitopes derived from endogenously synthesized antigens, i.e., upon mRNA delivery, and generate potent CD4+ T cell responses. 43 As opposed to MHC class I peptides, MHC class II-binding peptides are usually longer and more variable in length due to varying peptide residues on the N- and C-terminus centered around a core epitope.44–47 In this study, MHC class II-binding SpCas9-specific peptides were predicted using the IEDB prediction tool, in which each peptide was assigned a median consensus percentile rank representing its relative binding affinity to MHC class II (I-Ab). 34 A threshold rank equal to or lower than 6% was selected, yielding 45 peptides, whose sequences overlapped across nine distinct domains (Supplementary Table S2, underlined). To aid antigen presentation, flanking residues were added to the core of the overlapping peptides by extending the amino acid sequence on the N- and/or C-terminus of the predicted peptide (Supplementary Table S2, not underlined). 46 The resulting nine polypeptides covering the binding cores of 45 predicted peptides were considered potential candidates for further investigation (Supplementary Table S2).
Identification of SpCas9 protein-specific epitopes for CD4+ and CD8+ T cell recognition
T cell recognition of the predicted SpCas9-specific MHC class I- and II-binding peptides can be assessed ex vivo by quantifying cytokine production, which serves as an indirect measurement of T cell activation. To screen for CD8+ T cell responses, lymph node-derived cells recovered from immunized mice (Fig. 1C) were pooled and restimulated with each SpCas9-specific H2-Kb or H2-Db peptide, a positive control peptide (OVA), or left unstimulated for 6 h in total. Brefeldin A was added to all conditions for the last 4 h to allow the detection of intracellular cytokines using flow cytometry, and gated according to the gating strategy (Supplementary Fig. S3). Around 7% of CD8+ T cells stained positive for IFN-γ upon stimulation with pOVA, and several SpCas9-specific peptides induced CD8+ T cell activation above the background levels (medium only) (Fig. 2A,B). Particularly, six spCas9-specific peptides gave rise to ∼2–4% IFN-γ+ CD8+ T cells: five predicted H2-Kb binders (positions 880, 209, 447, 1043, and 1322) and one H2-Db binder (804) (Fig. 2B). Upon restimulation of lymph node cells derived from untreated mice (control group), these peptides did not induce T cell activation (Supplementary Fig. S4).

SpCas9-derived epitope identification for CD4+ and CD8+ T cell recognition.
To identify MHC class II-binding epitopes recognized by CD4+ T cells, splenocytes were restimulated with control OVA protein, SpCas9 protein, SpCas9-specific MHC class II-binding polypeptides, or left unstimulated for 3 days, before quantifying the IFN-γ concentration in the supernatants. The highest IFN-γ release was induced by OVA, followed by SpCas9 protein and pCas9 I-Ab 438 (Fig. 2C). While nonsignificant, three other peptides—pCas9 I-Ab 1121, 971, and 1005—also induced elevated IFN-γ secretion. Interestingly, the polypeptides eliciting IFN-γ secretion above background have all been predicted with a more strict criterion (%Rank ≤2) than the one we selected (%Rank ≤6), suggesting a successful epitope discovery by the algorithm (not shown).
Detection of SpCas9-directed immune responses upon repeated liver-targeting LNP administration
The induction of SpCas9-directed immune responses with nonviral delivery systems such as LNPs in mouse models has not yet been reported. Therefore, we investigated whether repeated administrations of nonadjuvanted, hepatocyte-targeting LNPs induces detectable SpCas9-specific immune responses. We formulated DLin-MC3-DMA-based LNPs co-encapsulating SpCas9-NLS mRNA and CCR5-targeting sgRNA (1:2, w/w) (Fig. 3A). The resulting LNPs exhibited a diameter of ∼105 nm, low polydispersity (0.09), and an encapsulation efficiency of ∼95% (Fig. 3B), as confirmed by visualizing the RNA using a gel retardation assay (Supplementary Fig. S2). The LNPs were administered IV in three doses given 1 week apart at a relatively low dose of 10 μg of total RNA (0.5 mg/kg; one-third corresponding to SpCas9 mRNA) (Fig. 3C). Six days after the final administration, splenocytes were restimulated for 3 days in the presence or absence of SpCas9 protein. T cell activation was measured indirectly by detecting the IFN-γ levels in the supernatant using ELISA. SpCas9 stimulation resulted in significantly elevated IFN-γ levels compared to untreated control (Fig. 3D).
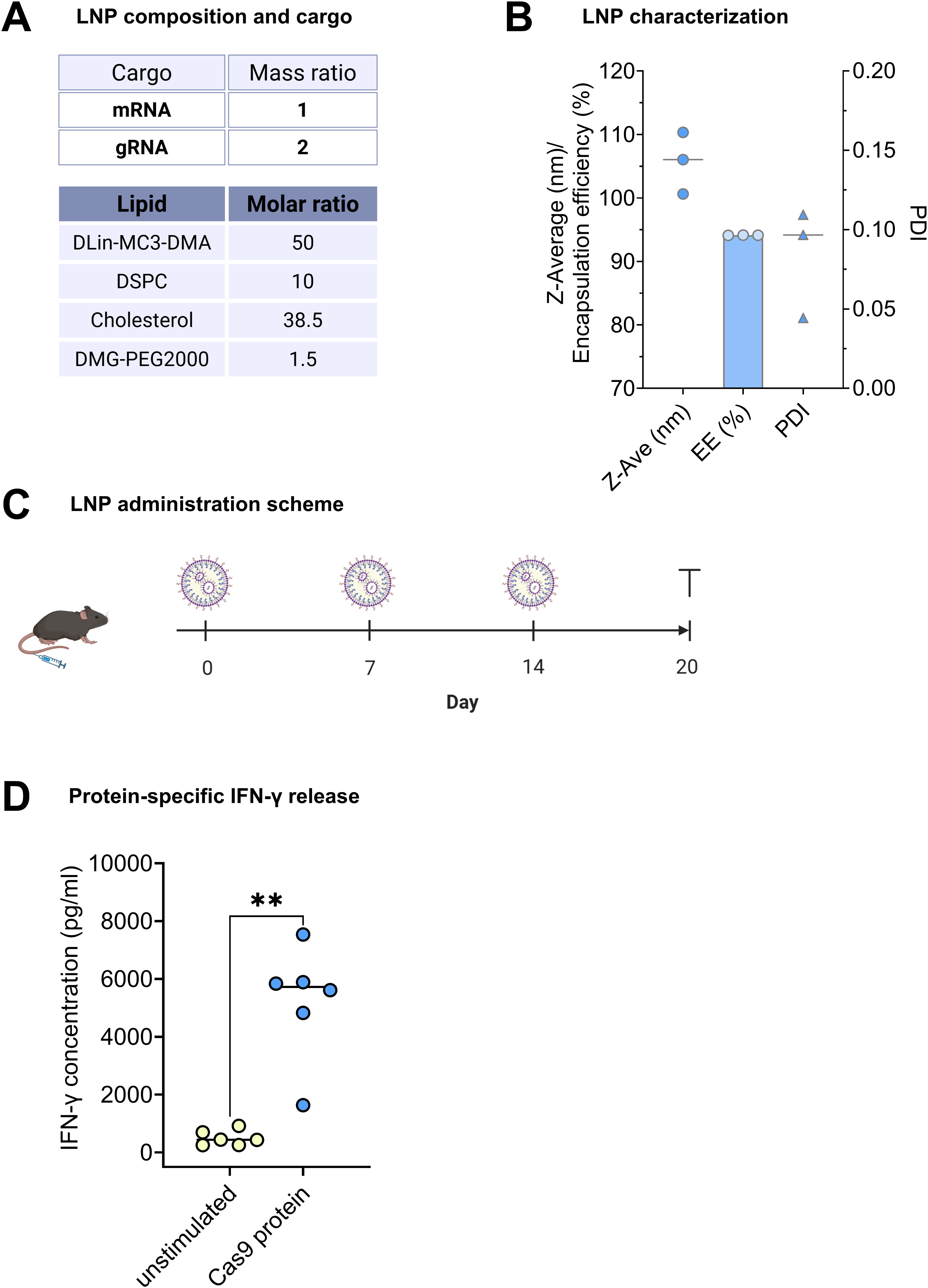
Induction of SpCas9-directed responses after systemic administration of a gene-editing LNP formulation.
Detection of anti-Cas9 antibody response upon LNP administration
SpCas9-specific antibody levels have been used as a measure of SpCas9-specific immunity induced in preclinical models upon gene therapy.9,13,19 We evaluated anti-SpCas9 IgG1 antibody levels upon repeated administration of adjuvanted or liver-targeting LNPs, as well as in control mice that received IV PBS injections at the same time points. Antibody levels on days 14 and 20 were considered significant if the background-subtracted OD value was higher than three standard deviations above the mean value on day 0. Using this threshold, antibodies were detected in 1 out of 6 and 3 out of 6 mice from the adjuvanted LNP group on days 14 and 20, respectively (Fig. 4A). Only 1 out of 6 mice developed an above-background response on days 14 and 20 in the liver-targeting LNP group. Thus, the average antibody response was higher upon immunization with adjuvanted LNPs compared to liver-targeting LNPs, which can also be observed by comparing normalized OD values (Fig. 4B). Of note, SpCas9-directed antibodies were in all cases only detected after the second LNP injection, which is in line with undetectable SpCas9-directed immunity before LNP-mediated exposure to SpCas9.
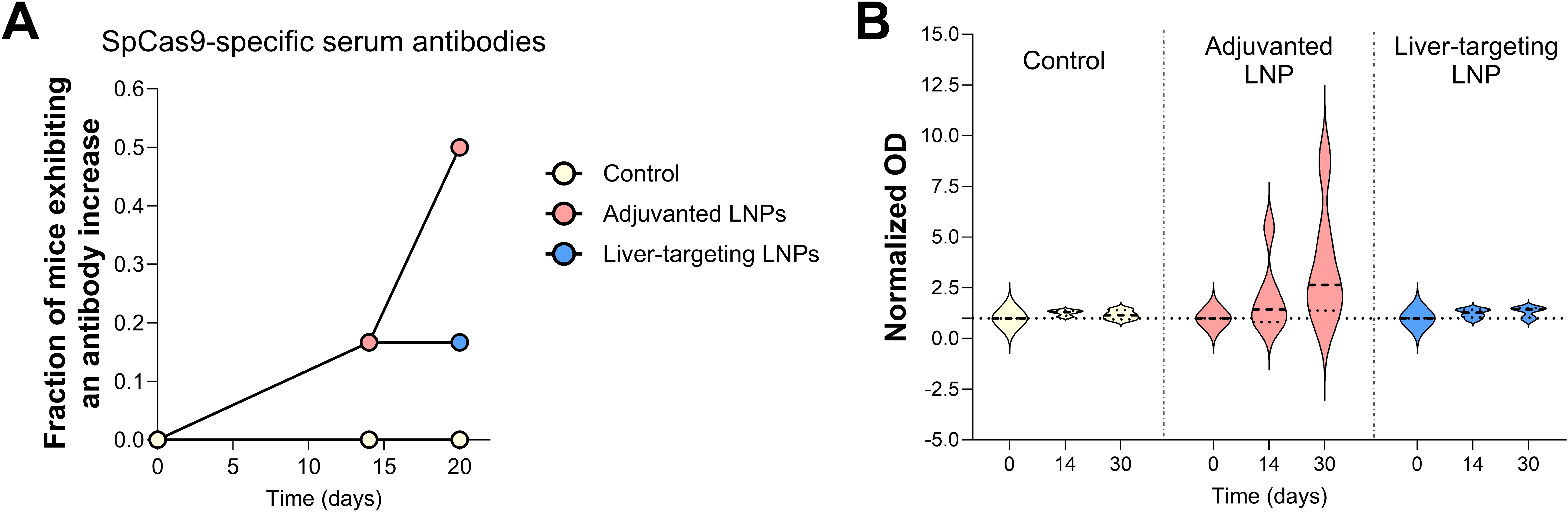
The increase in antibody levels upon repeated LNP administration.
Discussion
In this work, we assessed immune responses evoked against the SpCas9 nuclease, one of many SpCas9 variants originating from prokaryotes. Cas9 immunogenicity in humans has been recognized as a possible safety concern in gene therapies; however, only a handful of studies explore Cas9-specific immunity in detail. Preclinical studies commonly assess the tolerability of a gene therapy by measuring Cas9-specific serum antibodies,14,19,24,48 elevations in serum enzymes as markers of liver toxicity,14,19,49 or serum cytokine levels.16,49 While these approaches can provide insight into B cell-derived adaptive immunity and treatment-associated toxicity, respectively, they do not provide critical information on T cell activity. Specifically, the induction of cytotoxic T cells following intracellular Cas9 expression is particularly relevant, as Cas9 delivery vectors, including LNPs, can trigger inflammatory responses. It has been shown before that Cas9-directed cytotoxic T cell responses can lead to the elimination of edited cells through recognition of Cas9-derived epitopes on the surface of edited cells23,50 and tissue inflammation. 13 Therefore, identifying CD4+ and CD8+ T cell epitopes would be informative to evaluate responses in mouse models upon gene therapy. Moreno et al. found low SaCas9-specific, but no detectable SpCas9-specific, CD4+ T cell responses against predicted MHC class II peptides in the C57BL/6J model using an IFN-γ ELISPOT assay. 50 The same study also showed in vivo clearance of splenocytes pulsed with pooled SpCas9-derived MHC class I peptides in immunized animals. Nevertheless, the information on epitope dominance in this model has not been reported, thereby hindering the detection of epitope-specific cytotoxic T cells.
Mice raised under specific pathogen-free conditions do not exhibit detectable SpCas9-specific T cell responses28,50 or antibodies.9,19 To identify SpCas9-derived epitopes that can be recognized by T cells, mice were immunized IM with adjuvanted LNPs. Besides SpCas9 mRNA, the LNPs co-encapsulated mRNA coding for a commonly used model antigen, OVA, allowing the detection of antigen-specific CD8+ T cell responses. To improve antigen presentation, the open reading frames of the mRNA molecules were modified by flanking the antigen-coding sequences with domains that enhance antigen trafficking to antigen-processing compartments.35,36,38 OVA-coding mRNA molecules transfected into BMDCs resulted in detectable antigen presentation, as measured using the reporter B3Z T cell line. Next, the adjuvant poly(I:C) was included in the LNPs to additionally boost CD8+ and CD4+ T cell responses, thus increasing the chances for epitope identification.51–54 Poly(I:C) is a dsRNA analog and is therefore recognized by intracellular and endosomal RNA virus-sensing receptors such as RIG-I 55 and TLR3. 56 This recognition induces not only proinflammatory cytokine expression but also a cellular antiviral response, resulting in translation inhibition.57,58 Poly(I:C) concentration-dependent decrease in protein translation has been observed before. 53 Using in vitro assays, we defined 1:500 as the optimal ratio between poly(I:C) and total mRNA, resulting in dendritic cell co-stimulation as well as antigen recognition by CD8+ T cells. Comparable mRNA:poly(I:C) ratio in polymeric nanoparticles enabled the induction of antigen-specific CD8+ T cells, confirming our results. 53
Adjuvanted poly(I:C) LNPs were subsequently used for the induction of strong immune responses in the C57BL/6 mice, a commonly used (reporter) mouse strain for gene editing. 59 The LNPs co-encapsulated both SpCas9 and OVA mRNA (1:1 molar ratio). OVA mRNA was included as a positive control for induced immune responses for comparison with SpCas9. Repeated IM administration of the LNPs facilitated the induction of SpCas9- and OVA-specific immunity, as measured by total IFN-γ production (Fig. 1E), cytotoxic T cells (Fig. 1D), and the presence of anti-Cas9 antibody in 50% of the animals (Fig. 4). These results confirm the de novo induction of SpCas9-directed cell-mediated immunity, allowing us to screen for SpCas9-derived epitopes for CD8+ and CD4+ T cell recognition.
Peptide screening for CD8+ T cell epitopes on lymph node cells yielded six predicted SpCas9-derived MHC class I-binding peptides, scattered across different SpCas9 domains that induced at least 2% IFN-γ-expressing CD8+ T cells (pCas9-Kb 209, KAILSARL; pCas9-Kb 447, RIPYYVGPL; pCas9-Db 804, TQLQNEKLYL; pCas9-Kb 880, KNYWRQLL; pCas9-Kb 1043, MNFFKTEI; pCas9-Kb 1322, AAFKYFDTTI). Predicted binding affinities for their corresponding MHC class I molecules range between 37 and 148 nM (Supplementary Table S1), while OVA-derived peptide SIINFEKL exhibits a superior affinity for H2-Kb of 10 nM. In line with this, the model OVA-derived peptide yielded a response of 7.5% CD8+ T cell activation, which was higher than the highest-scoring SpCas9-derived peptide 880 (predicted affinity of 37 nM). Next, CD4+ T cell epitope screening on splenocytes with predicted MHC class II-binding polypeptides detected four that induced above-background IFN-γ responses. Among them, polypeptide pCas9 I-Ab 438 (EKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEE) induced a statistically significant (p = 0.01) increase in IFN-γ compared to the untreated control. Interestingly, this sequence covers a CD8+ T cell epitope pCas9-Kb 447, potentially explaining the superior T cell activation.60,61
Finally, we investigated SpCas9-directed immune responses in a therapeutically relevant setting. We formulated hepatocyte-targeting LNPs encapsulating mRNA for repeated systemic administration in mice. Despite their lower immunostimulatory profile compared to viral vectors, LNPs can induce low-level inflammation due to their ionizable lipid components. 32 We performed an extracellular protein challenge ex vivo, which predominantly stimulates CD4+ T cells. All mice developed statistically significant SpCas9-specific T cell responses compared to unstimulated control (Fig. 3D), confirming the induction of SpCas9-directed immunogenicity using a nonviral drug delivery system. The induction of CD4+ T cell responses upon mRNA delivery using LNPs can be explained by APC-mediated uptake of LNPs and presentation of endogenously synthesized antigen. 43 However, resolving the relative contributions of CD4+ and CD8+ T cell subsets to cytokine release would have been more informative than the bulk cytokine measurements performed in this study. Low or undetectable anti-Cas9 antibody levels were measured, in line with previously published data, 19 whereas adjuvanted LNPs induced stronger humoral immunity (Fig. 4). The absence of Cas9-specific antibodies upon LNP delivery is not unexpected, since SpCas9 is not secreted or otherwise available for extracellular uptake by B cells. This contrasts with naturally established preexisting immunity in humans, where a correlation between Cas9-specific T cell and antibody responses was observed. 6 The discrepancy between cellular and humoral immune responses further supports the need to monitor Cas9-specific immune responses on the T cell level. Such monitoring could prove valuable for preclinical risk assessment of immunogenicity of CRISPR/Cas9-based therapeutic products.
The main limitation of this study is the relevance of translating the immunological outcomes to humans. Because S. pyogenes is primarily a human-specific pathogen, 62 neither infection of mice with Cas9-bearing bacteria nor immunization with SpCas9 is sufficient to fully recapitulate immunity in humans. Still, preclinical testing of novel drug formulations is mostly performed in mouse models, and so far, measuring (cytotoxic) T cell responses has been overlooked. Nonetheless, the few studies that did dive into the immunological events upon gene therapy found striking results, further confirming the importance of immune monitoring.13,23,50
Conclusion
Our findings confirm the de novo induction and detection of SpCas9-specific cell-mediated immunity upon repeated nonviral SpCas9 mRNA delivery with LNPs and the discrepancy between T cell and antibody responses. Considering that de novo SpCas9-specific immunity may be induced in naive mice upon repeated liver-targeted LNP therapy, Cas9 expression could trigger reactivation of existing Cas9-specific T cells in humans. This study identifies six MHC class I (H-2Kb/d)-binding SpCas9-derived peptides that stimulate CD8+ T cells, as well as one polypeptide with MHC and class II (I-Ab)-binding sequences that induced strong CD4+ T cell activation in C57BL/6 mice. We further found that pCas9 I-A438 polypeptide includes a sequence of a CD8+ T cell dominant epitope, pCas9-Kb 477, which likely contributes to the strong T cell activation. This specific polypeptide could be used for monitoring CD4+ T cell activation of effector or even regulatory T cells, which are important for Cas9-specific immune tolerance. 2 Moreover, developing immunosilent gene therapies that do not activate T cells would be ideal. In this context, T cell epitopes would enable monitoring of T cell responses in preclinical models of gene therapy, especially upon repeated dosing.
Authors’ Contributions
D.P.: Data curation, formal analysis, investigation, methodology, project administration, visualization, and writing—original draft preparation. N.B.: Investigation, supervision, and writing—review and editing. A.S.: Supervision and writing—review and editing. F.B.: Supervision and writing—review and editing. E.M.: Conceptualization, funding acquisition, supervision, and writing—review and editing.
Footnotes
Acknowledgment
The authors thank the Flow Cytometry and Cell Sorting Facility at the Faculty of Veterinary Medicine at Utrecht University for support.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by the Netherlands Organization for Scientific Research Talent Program VICI (grant number
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.