
Abstract
Craniosynostosis is a rare congenital bone condition where skull sutures fuse prematurely and is linked to mutations in over 60 genes. Generating mutation-specific in vitro models allows investigation of craniosynostosis-associated mutations without the need for patient-derived material or transgenic gene expression. Here, we developed a human in vitro disease model with the CRISPR-Cas9 prime editing variant, using an immortalized TERT-immortalized mesenchymal bone marrow-derived stem (MSC-TERT) cell line with osteogenic potential. MSC-TERT cells showed a higher resistance to prime editing, compared with HEK293FT cells. Addition of dnMLH1 and epegRNAs resulted in higher editing efficiencies in HEK293FT cells, but not in MSC-TERT cells. Prime editing efficiency varied between targeted loci and was found to be more efficient in nonadherent cells compared with adherent cells. Prime editing continued over 4 days in an isolated nonadherent HEK293FT culture. Our results present a foundation on the use of prime editing to establish FGFR2 mutation-specific in vitro models and their application in MSC-TERT cells.
Introduction
Craniosynostosis is a rare congenital bone condition in which one or more sutures between the calvarial plates ossify and fuse prematurely.1,2 During healthy development, the ossification process occurs gradually over time postnatally and the sutures do not fuse completely until adult age. 3 In craniosynostosis, however, the affected suture fuses during the development of the fetus and within the first year after birth. While nonaffected sutures allow separation and flexibility between the calvarial plates, for example, facilitating both childbirth and postnatal brain growth, premature fusion restricts the skull from expanding in tandem with the developing brain.1–4 Infants with craniosynostosis typically present with abnormal skull morphology and are at risk for elevated intracranial pressure. 5
Craniosynostosis can arise from environmental risk factors or from genetic mutations. 6 Genetically, craniosynostosis is considered highly multigenic, with mutations identified in over 60 different genes.7,8 Mutations in one of the more frequently affected genes, fibroblast growth factor receptor 2 (FGFR2), are associated with a multitude of phenotypically distinct syndromes, such as Crouzon, Apert, Pfeifer, and Beare–Stevenson cutis gyrata.9,10 In many cases, bilateral coronal suture synostosis or fusion of multiple sutures is observed. 11 Many of these syndromes are commonly associated with a range of potential causative genetic mutations. This genetic heterogeneity presents a significant challenge for both research and clinical diagnosis of craniosynostosis, as there is no single, clear genetic target. Moreover, the various mutations often impact protein function in distinct ways. To effectively study the diverse genotypic causes of craniosynostosis and to systematically compare the effects on FGFR2 function, there is a strong need for easily customizable, mutation-specific human cell models developed in vitro.
Research on genetic craniosynostosis largely depends on the use of nonhuman models in which cellular and molecular interactions and mechanisms have been investigated.12–16 Alternatively, to investigate the cellular interactions and molecular mechanisms involved in craniosynostosis, in vitro models that make use of transgenic overexpression of the mutation-of-interest are commonly utilized.17–21 Unfortunately, both of these approaches do not fully recapitulate the condition as presented in patients, either due to not being human-derived or intervening with the endogenous expression of the alleles, respectively. Patient-derived cell lines usually provide an excellent solution to these issues. However, in cases where mutations are exceedingly rare, or causative mutations are highly multigeneic, it is not feasible or perhaps near impossible to model all of those mutations. On top of that, the patient-derived lines will be primary cell cultures that pose certain challenges. Such challenges include the risk of heterogenous cell populations, a finite lifespan that ends in senescence, and the populations may differ in performance to a great extent from each other due to donor variances.22–25 Although donor variance might play an important role in personalized medicine by taking individual differences into account, it defeats the purpose of having a standardized, consistent cell line that eliminates many confounders. Therefore, a well-described, standardized, and continuous cell line would make a more appropriate fit to model many different mutations in such a fashion. Taken all of this together, the ideal solution consists of a tissue-specific, immortalized cell line in which it is able to efficiently introduce any mutation of interest, without the need for patient-derived material or transgenic overexpression. To this end, we explored the possibilities of creating a human in vitro disease model for the craniosynostosis-associated FGFR2-C342Y Crouzon mutation in TERT-immortalized mesenchymal bone marrow-derived stem cells (MSC-TERT cells) using CRISPR-Cas9 prime editing. 26
Prime editing is a variant of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 gene editing that uses a nickase Cas9(H840A) fused with a Moloney Murine Leukemia Virus Reverse Transcriptase (RT), and an adjusted prime editing guide RNA (pegRNA) that doubles as a template to precisely rewrite small portions (up to 60 bp) of the target genome.27,28 In contrast to other editing techniques, prime editing can introduce small insertions or deletions as well as substitutions without generating double-stranded DNA breaks (DSBs). Recently, several adjustments and optimizations of the system have resulted in enhanced prime editing efficiency. Such optimizations include the addition of a secondary nicking short guide RNA (ngRNA) that targets the complementary strand of the pegRNA (termed PE3), the addition of a dominant negative variant of MutL Homolog 1 (dnMLH1) (termed PE5), the use of an engineered pegRNA (epegRNA), and codon optimization of the prime-editor Cas9 (PEmax-Cas9).27,29,30
The objective of the current study is to use optimized prime editing protocols to generate in vitro disease models for FGFR2-linked craniosynostosis mutations in MSC-TERT cells. This study presents results that provide a fundamental basis for introducing gene mutations responsible for a wide range of rare bone diseases.
Materials and Methods
Cloning
The custom pegRNA constructs are synthesized by cloning in the spacer-, scaffold-, RT template (RTT)-, and primer binding site (PBS)-sequences (Supplementary Table S2) inside the pU6-pegRNA-GG-acceptor plasmid by HiFi DNA Assembly (New England Biolabs; #E5520S). Single-stranded (ss) oligonucleotides are ordered, annealed, and assembled in an open pU6-pegRNA-GG-acceptor backbone. After bacterial expansion, the plasmid constructs are isolated and subsequently used in the prime editing experiments.
Oligonucleotide annealing
Ss oligonucleotides are diluted to a concentration of 100 µM and annealed using the following protocol adapted from Anzalone et al. 27 :
For one reaction, 1 µL of top oligonucleotide (100 µM) and 1 µL of bottom oligonucleotide (100 µM) are mixed with 23 µL
The annealed oligonucleotide product (4 µM) is diluted 1:4 with nuclease-free water to a stock concentration of 1 µM. The stock concentration is diluted 1:10 for a final working concentration of 0.1 µM (0.1 pmol).
Nuclease-free water 10 mM Tris–Cl with pH of 8.5 50 mM NaCl
Backbone digestion and purification
The pU6-pegRNA-GG-acceptor (backbone) plasmid is digested by BsaI. The digestion product is run on a 1% Agarose gel for electrophoresis. The digested plasmid fragment at ∼2.2 kb is isolated and purified with the PureLink Gel extraction kit (Invitrogen; #K210025) by following the manufacturer’s protocol. The concentration of the purified, digested backbone plasmid is measured by Nanodrop and subsequently used for HiFi DNA assembly.
Hybridization and ligation
HiFi DNA assembly
Following HiFi DNA Assembly guidelines, the spacer fragment and 3′ extension fragment (RTT + PBS sequence) were designed with 20-nucleotide overlaps on both sides with the adjacent fragment or backbone sequence. The scaffold fragment and the backbone sequences are overlapped by the 20-nucleotide overlaps from the other two fragments. Supplementary Table S2 lists which spacer-, RTT-, PBS-, and epeg linker-sequences have been used in the design and assembly of the (e)pegRNA constructs.
When designing fragments for the secondary nicking guide, both the spacer fragment and the scaffold fragment overlap each other as well as the backbone sequence. Supplementary Table S3 lists the spacer sequences that have been used in the design and assembly of the (secondary nicking) sgRNA constructs.
HiFi DNA assembly (New England Biolabs; #E5520S) was performed following the manufacturer’s protocol on “4–6 fragment Assembly” for (e)pegRNA constructs and “2–3 fragment Assembly” for (secondary nicking) sgRNA constructs.
The assembly mixtures were incubated at 50°C for 60 min, after which the mixtures were subsequently used for bacterial transformation.
sgRNA design
The spacer sequence of the secondary nicking sgRNAs has been selected in the Santa Cruz Genome Browser. We selected sgRNA spacer sequences with a high predicted cleave value (Doench 2016 score >55%; preferably >85%) and a low expectation of off-target effects (MIT score >50).
Bacterial transformation
Plasmid constructs are expanded in NEB DH5α competent Escherichia coli bacterial cultures (New England Biolabs; #C2987). The NEB DH5α cells were chemically transformed following the manufacturer’s protocol with some adjustments. The applied protocol is detailed as follows: NEB 5-alpha Competent E. coli cells were thawed on ice. Then, 5 µL of the Kinase, Ligase, and DpnI (KLD)-treated plasmid mix was added to the thawed cells. The plasmid-cell mix was placed on ice for 30 min. The mix was heat shocked at 42°C for 30 s and afterward placed on ice for 5 min. Then, 250 µL of room temperature Super-Optimal broth with Catabolite repression (SOC) medium is added to the mixture, and the mixture is subsequently incubated at 37°C for 60 min on a platform shaker (250 rotations per minute [rpm]). From the mixture, 25 µL and 225 µL is spread onto separate selection plates and incubated overnight at 37°C.
Bacterial expansion
Isolated colonies from the selection plates were picked and incubated overnight at 37°C in 5 mL of 2-YT Broth (Invitrogen; #22712020) (mini prep) supplemented with 100 µg/mL Ampicillin (Sigma-Aldrich; #A9518) on a platform shaker going 250 rpm. Afterward, 100 µL of the overnight culture is transferred to 100 mL of 2-YT Broth (midi prep) supplemented with 200 µg/mL Ampicillin and incubated overnight at 37°C on a platform shaker going 250 rpm.
DNA isolations
Plasmid isolation
After bacterial expansion, the mini prep plasmid DNA was isolated with the PureLink Quick Plasmid Miniprep Kit (Invitrogen; #K210011) following the manufacturer’s protocol. The midi prep plasmid DNA was isolated with the QIAGEN Plasmid Midi Kit (QIAGEN; #12143) following the manufacturer’s protocol.
DNA isolation from attached culture
Cell cultures have their medium aspirated, and are washed once with 1× Dulbecco’s phosphate buffered saline (1× DPBS). Genomic DNA (gDNA) is isolated from the cells using ∼20 µL/cm2
DNA isolation from floating cell fraction
Culture medium containing the floating cell fraction (FCF) is separated from the attached cells. The separated medium is centrifuged at 500 ×g for 5 min. The supernatant is aspirated, and the cell pellet is washed once with 1× DPBS. Then, the cells are centrifuged once more at 500 ×g for 5 min. The supernatant is aspirated, and the cell pellet is dissolved in
Lysis buffer
Lysis buffer fit for DNA isolation is synthesized by supplementing
50 mM KCl 10 mM Tris–HCl (pH 8.3) 2.5 mM MgCl2 0.1 mg/mL gelatin (Sigma-Aldrich; #73865) 0.45% (v/v) NP-40 Alternative (Sigma-Aldrich; #492018) 0.45% (v/v) Tween20 (Sigma-Aldrich; #P1379) Autoclave to sterilize and dissolve gelatin
Cell culture
HEK293FT culture
HEK293FT cell lines were seeded with 1.5 × 106 cells in a T-175 culture flask (∼8.6 × 103 cells/cm2), and cultured with Dulbecco's Modified Eagle Medium (DMEM) culture medium (Gibco; #11965092) supplemented with 10% v/v fetal bovine serum (FBS) (Gibco; #A5670401), 2% v/v Penicillin/Streptomycin (P/S) (100 U/mL of Penicillin and 100 U/mL Streptomycin) (Gibco; #15140148). The HEK293FT cells are cultured until they reach 80–90% confluency. During passaging, the cells are washed once with 1× DPBS, and incubated with 0.5% Trypsin-Ethylenediaminetetraacetic acid (EDTA) (1:10 diluted with 1× DPBS) (Gibco; #15400054) for 5 min at 37°C.
MSC-TERT culture
MSC-TERT cell lines are seeded with 0.5 × 106 cells in a T-175 culture flask (∼3.0 × 103 cells/cm2) and cultured with custom phenol red-free, calcium chloride-free αMEM (Gibco), supplemented with 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Sigma-Aldrich; #H3375), 1.8 mM CaCl2, 10% v/v FBS (Gibco; #A5670401), 2% v/v P/S (100 U/mL of Penicillin and 100 U/mL Streptomycin) (Gibco; #15140148), and the pH set to 7.5. The MSC-TERT(Cas9) cells that stable express endogenous spCas9 are, 24 h after seeding, selected for their Cas9-expression with 2 µg/mL Puromycin (Sigma-Aldrich; #P7255) for 48 h. The MSC-TERT and MSC-TERT(Cas9) cells are cultured until they reach 80–90% confluency. During passaging, the cells are washed once with 1× DPBS, and incubated with 0.05% Trypsin-EDTA (1:10 diluted with 1× DPBS; Gibco; #15400054) for 5 min at 37°C.
Transfection
Transfections were performed with the P2 Primary Cell 4-D Nucleofector X Kit S (Lonza; #V4XP-2032). Standard manufacturer’s protocol was followed when performing the transfections. For both the HEK293FT and MSC-TERT cells, 200,000 cells per condition were used. During the green fluorescent protein (GFP) transfection efficiency experiments, HEK293FT and MSC-TERT cells were transfected with 1500 ng pCMV-PE2-P2A-GFP each. HEK293FT cells were cultured for 24 h post-transfection at 37°C until processed for FACS analysis. Five separate MSC-TERT transfections were performed in parallel. The MSC-TERT cells were cultured for the duration of their respective days post-transfection at 37°C until processed for FACS analysis. HEK293FT cells were transfected by with the Nucleofection program CM-130, and the MSC-TERT cells with the Nucleofection program EH-100.
During the prime editing experiments, HEK293FT and MSC-TERT cells are transfected with 500 ng (e)pegRNA plasmid (Supplementary Table S2), 1500 ng pCMV-PE2 or pCMV-PEmax-P2A-BSD, and, if applied, 167 ng of secondary nicking sgRNA (Supplementary Table S3) and 750 ng pEF1a-hMLH1dn. The cell cultures are kept in culture for 5 days post-transfection until processed for DNA isolation. The cell cultures are refreshed after 48 h in culture. During the experiments with pCMV-PEmax-P2A-BSD, the cultures are refreshed after 24 h post-transfection and put on Blasticidin (Thermo Fisher Scientific; #R21001) selection medium (5 µg/mL for HEK293FT cells and 10 µg/mL for MSC-TERT cells) for 24 h and afterward refreshed after 48 h.
During the experiments with the CRISPR-Cas9 FGFR2 knockout constructs, the HEK293FTC342Y/+/+/+ cells are transfected with 1.5 µg Cas9 plasmid and 500 ng FGFR2-ex7-KO sgRNA plasmid. Otherwise, they follow the same transfection and culturing schedule as the other conditions.
During the indel generation in FGFR2 experiments, the MSC-TERT(Cas9) cells are transfected with 4 µg of FGFR2(C342Y) or HEK3(CTTins) pegRNA plasmid. The cells are cultured for 5 days post-transfection, and refreshed every 48 h, until processed for DNA isolation.
PCR
Regions of interest were amplified by Polymerase Chain Reaction (PCR). When using crude gDNA templates in the PCR, 1–3 µL of DNA template was added to the reaction mix. PCR was performed using the reaction setup and thermocycler Hot Start PCR program from the manufacturer’s protocols (Promega [G2 Hot Start Polymerase]).
Sanger sequencing
Samples sent for Sanger sequencing were prepared by mixing 5 µL of PCR product (5 ng/µL for 300–1000 bp amplicons; 10 ng/µL 1000–3000 bp amplicons) with 5 µL primer (5 µM) and sent to Eurofins Genomics for LightRun Tube analysis.
Next-generation sequencing
Samples sent for next-generation sequencing (NGS) were generated by amplifying the region of interest by TrueSeq Adaptor primers detailed in Supplementary Table S5. The PCR product is purified with the PureLink PCR purification kit (Invitrogen; #K310001) and sent to Eurofins Genomics following their short-read amplicon sequencing using the Illumina submission protocol. NGS data were analyzed with CRISPRESSO2. 31
TIDE analysis
Quantification of prime editing from the HEK3(CTTins) construct and the indel generation in the HEK3 site and FGFR2 were performed with TIDE analysis. 32 Control sample chromatogram files consisted of the untransfected control conditions from each of the experiments.
Restriction enzyme digestion
The restriction enzyme digestions were performed by amplifying the region of interest by PCR and purifying the PCR product with the PureLink PCR purification kit (Invitrogen; #K310001). For each reaction, 500 ng of PCR product is mixed with 5 units of the respective restriction enzyme (New England Biolabs: RsaI [#R0167]; BstUI [#R0518]; SmaI [#R0141]), 10% v/v rCutsmart buffer (New England Biolabs), and RNAse-free water (Promega; #MC1191) up to 30 µL.
Band intensity calculations
The intensity of PCR bands in images derived from gel electrophoresis was done with FIJI (Fiji Is Just ImageJ). The gel electrophoresis image file is opened in FIJI in grayscale. The 600 bp band was used for measurement in all samples. Band intensity was determined by calculating the gray area and normalizing the intensity to the Day 1 sample.
Flow cytometry
GFP content in cells was measured by flow cytometry 24 h post-transfection. Supernatant containing floating cells was collected. The cell culture is washed once with 1× DPBS and subsequently incubated with 0.5% Trypsin-EDTA (1:10 diluted with 1× DPBS; Gibco; #15400054) for 5 min at 37°C. The cell suspension is collected and, together with the supernatant, passed through a 72 µm sieve. GFP-positivity is detected with a 488 nm laser channel and plotted against the same channel with a different filter to compensate for autofluorescence.
The HEK293FTC342Y/+/+/+ cell lines were derived from flow cytometry-assisted cell sorting. The FGFR2(C342Y) + 62 prime-edited cell culture was washed once with 1× DPBS and subsequently incubated with 0.5% Trypsin-EDTA (1:10 diluted with 1× DPBS) (Gibco; #15400054) for 5 min at 37°C. The cell suspension is collected and passed through a 72 µm sieve. Singe cells were sorted in a new culture vessel. After recovery and expansion, the cell cultures were genotyped for the FGFR2-C342Y mutation by PCR and restriction enzyme analysis. Positive samples were confirmed with Sanger sequencing and NGS.
Results
Validation of transfection and prime editing efficiency in HEK293FT and MSC-TERT cells
To assess whether we could reproduce one of the mutations shown in the original publication on prime editing, 27 we used HEK293FT and MSC-TERT cells and targeted the HEK3 locus on chromosome 1. The pegRNA introduces a 3-base pair (bp) CTT segment into the genome, which we termed HEK3(CTTins).
We first transfected both cell lines with only a GFP coexpressing PE-Cas9 plasmid (CMV-PE2-P2A-GFP; hereafter termed PE-Cas9-GFP) to monitor transfection efficiencies. This resulted in satisfactory transfection efficiencies in both HEK293FT (58.8%) and MSC-TERT cultures (50.8%) (Fig. 1A and B). Using flow cytometry, we observed a noticeable decrease in the percentage of detected events in the Live Cells gating for the PE-Cas9-GFP-transfected samples 24 h after transfection, compared with the untransfected samples (Supplementary Fig. S1). To monitor the presence of the PE-Cas9-GFP construct over time, we determined the number of GFP-positive cells in the MSC-TERT cultures over 5 days by flow cytometry. The percentage of GFP-positive cells declined from ∼50% 1 day after transfection to ∼0.5% 5 days after transfection. This indicates that by day 5 post-transfection, the GFP-coexpressing Cas9 plasmid is no longer present in the majority of the cells in culture (Fig. 1B).
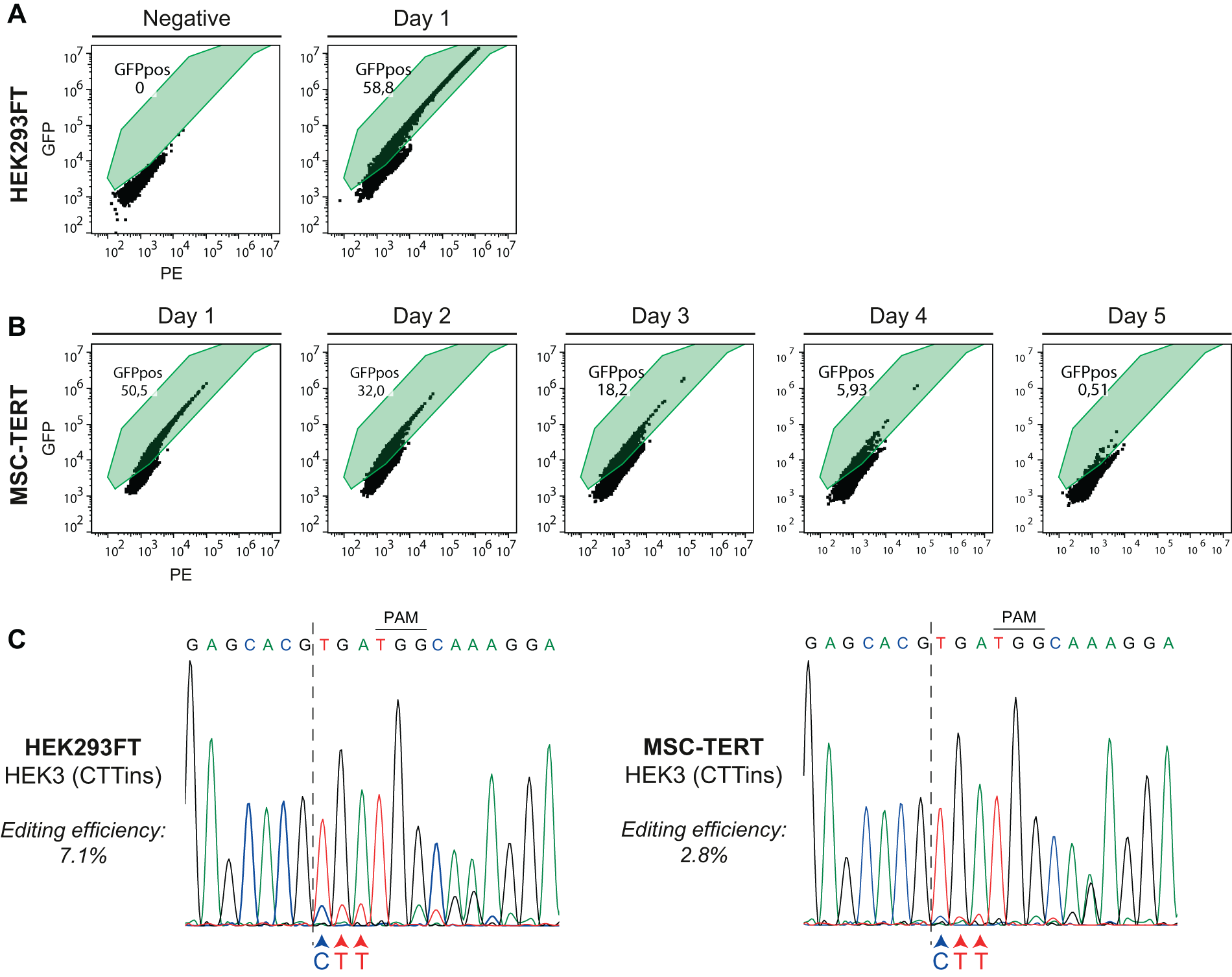
Transfection efficiency of lipofectamine and nucleofection plasmid delivery methods, and overview of prime editing in HEK3.
Next, HEK293FT and MSC-TERT cells were transfected with the HEK3(CTTins) pegRNA together with the PE-Cas9 plasmid. Sanger sequencing confirmed the incorporation of the CTT-insertion at the HEK3 locus in both HEK293FT and MSC-TERT cells; apart from overlapping signals due to the introduced CTT triplet, downstream overlapping signals are the result of a shift of these recombinant sequences (Fig. 1C). TIDE analysis of Sanger sequencing results showed an approximated prime editing efficiency up to 7.1% and 2.8% in HEK293FT and MSC-TERT cells, respectively (Supplementary Fig. S2). These data illustrate that our current setup is able to apply prime editing in MSC-TERT cells at the HEK3 site, albeit with a slightly lower prime editing efficiency as compared with the HEK293FT cells.
Targeting FGFR2 with prime editing yields incorporated mutations in HEK293FT but not in MSC-TERT cells
We next designed a custom pegRNA that targets exon 9 of FGFR2 and introduces the Crouzon-associated FGFR2-C342Y (c.1025 G > A; p.C342Y) mutation (Fig. 2A). Incorporation of the FGFR2-C342Y mutation creates an RsaI restriction site in the genomic sequence, enabling detection of prime editing success by restriction enzyme analysis. To prevent re-editing of novel prime-edited strands and increase editing efficiency,33,34 we simultaneously incorporate a synonymous mutation (c.1029 G > A; p.L343L), which disrupts the PAM site (Fig. 2A and B). In addition, to promote editing efficiency further, we designed two ngRNA constructs targeting PAM sites 33 bp and 62 bp downstream of the pegRNA nicking site; termed PE3 +33 ngRNA and PE3 +62 ngRNA, respectively.
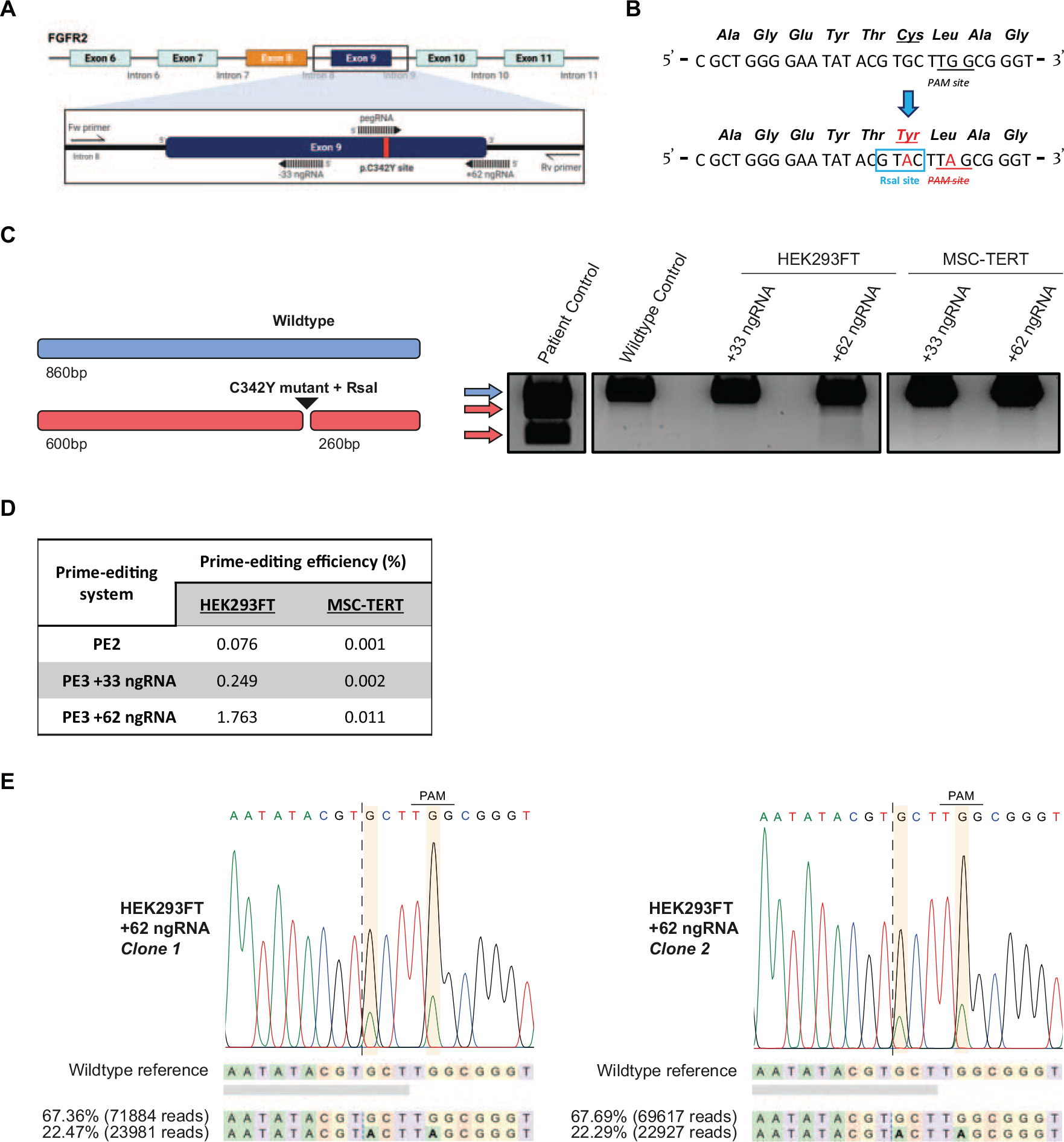
Overview of prime editing targeting FGFR2 and introduction of the C342Y mutation in HEK293FT and MSC-TERT cells.
An RsaI digested PCR product indicated the successful introduction of the Crouzon FGFR2-C342Y mutation in HEK293FT cells for the +62 ngRNA condition, but not for the +33 ngRNA condition (Fig. 2C). No digested PCR product was observed in the MSC-TERT cells either with the +62 ngRNA or with the +33 ngRNA conditions (Fig. 2C). NGS data showed varying prime editing efficiencies in HEK293FT cells depending on the ngRNA utilized (0.076% with no ngRNA; 0.249% with +33 ngRNA; and 1.763% with +62 ngRNA). MSC-TERT prime editing efficiencies were detectable, yet relatively low compared with HEK293FT cells, with all prime editing combinations used (0.001% using no ngRNA; 0.002% using +33 ngRNA; and 0.011% using +62 ngRNA) (Fig. 2D).
Similarly, when targeting fuzzy planar cell polarity protein (FUZ)–a recently reported craniosynostosis-associated mutation 35 – with prime editing, inclusion of a ngRNA resulted in a higher editing efficiency over that of the samples without a ngRNA (Supplementary Fig. S3). We observed the incorporation of the mutation in both HEK293FT and MSC-TERT cells. For all conditions, editing efficiencies in HEK293FT cells were higher compared with those in MSC-TERT cells. Moreover, the editing efficiency of FUZ was overall higher compared with that of FGFR2 (Supplementary Fig. S3). This suggests that MSC-TERT cells are susceptible to prime editing and that the prime editing efficiency may vary not only between cell types but also between loci.
Prime edited HEK293FT cells are tetraploid at edited FGFR2 locus
With the successful introduction of the FGFR2-C342Y mutation in the HEK293FT cells, we investigated whether we could derive a clonal cell line from this population. The HEK293FT cell population derived from the most efficient PE3 + 62 ngRNA set-up (hereafter termed “FGFR2(C342Y) + 62”) was expanded in culture and afterward single-cell sorted by flow cytometry cell sorting (FACS). Single-cell-expanded clones were analyzed by RsaI restriction enzyme digestion. Two clones were identified to harbor the FGFR2-C342Y mutation and were confirmed by Sanger sequencing (Fig. 2E).
Interestingly, we observed an unequal distribution of wild-type and recombinant genotypes in the Sanger sequencing data that may suggest that either our clonal lines were not derived from single cells or that the individual cells contain extra wild-type copies of FGFR2 (Fig. 2E). To exclude the possibility of having nonhomogenous sorted cell lines, both clonal lines were single-cell-sorted once more. All screened subclones harbored the C342Y mutation and also contained the same unequal distribution pattern of genotypes of the clonal line they were sorted from, confirmed by Sanger sequencing (Fig. S4A). NGS indicated that 22.47% of the sequencing reads consist of the mutant allele and 67.36% of the wild-type alleles in clone 1. For clone 2, similar percentages were obtained for the mutant (22.29%) and wild-type (67.69%) alleles (Fig. 2E; Fig. S4B). This indicates that our HEK293FT cells are likely tetraploid for the FGFR2 locus, and that one out of four alleles in these clonal lines harbor the intended C342Y mutation (we hereafter term these clonal cell lines: HEK293FTC342Y/+/+/+).
Optimization of prime editing parameters significantly improves FGFR2 editing efficiency in HEK293FT cells, but not in MSC-TERT cells
Despite our FGFR2(C342Y) + 62 setup having an editing efficiency capable of deriving clonal C342Y-mutant cell lines for HEK293FT cells, we failed to introduce the mutation in MSC-TERT cells. However, the absence of any successfully incorporated C342Y mutations in the MSC-TERT cells may still be a result of insufficient performance of our FGFR2(C342Y) + 62 constructs in a PE3 set-up in MSC-TERT cells. Therefore, we investigated whether various recently published adjustments for the prime editing system enable us to successfully introduce the FGFR2-C342Y mutation in MSC-TERT cells. These optimizations include (1) the addition of a dnMLH1 protein, (2) the use of an epegRNA instead of a pegRNA, and (3) using a codon-optimized PEmax-Cas9 protein29,30 (Supplementary Table S4).
The addition of the dnMLH1 construct to the PE3 system (termed PE5) improved the editing efficiency of FGFR2(C342Y) + 62 from the previously documented 1.8% to 5.4% in HEK293FT cells, and further increased up to 14.5% with the use of an epegRNA (termed ePE5). Despite PEmax-Cas9 being described as a codon-optimized construct that outperforms the PE-Cas9 construct, we observe a slightly lowered efficiency when using ePE5max (12.2%) (Fig. 3A). The MSC-TERT cells did not show any indication of successful incorporation of the intended mutation with any of these adjustments, despite showing improvement in HEK293FT cells (Fig. 3A).
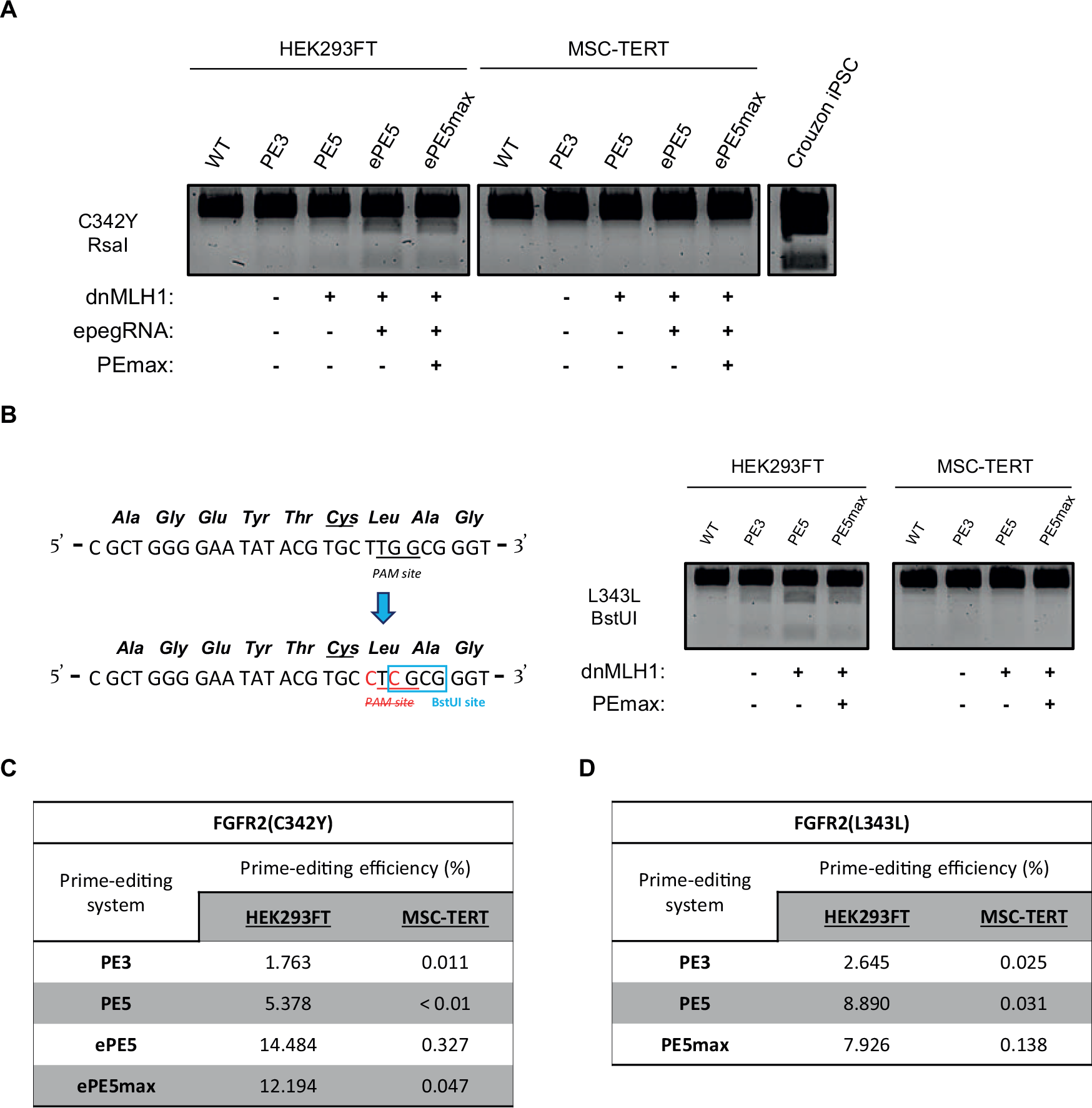
Overview of prime editing optimizations for the introduction of the C342Y- and L343L-mutation in FGFR2.
We considered the possibility that the prime editing constructs cannot interact with the DNA on our target site. To investigate this, we assessed whether random insertions and deletions (indels) are generated in MSC-TERT expressing conventional spCas9 following transfection with a pegRNA targeting FGFR2. We successfully detected indels at the target site, indicating that the FGFR2 locus is amenable to CRISPR-based editing (Supplementary Fig. S5).
Prime editing of HEK293FT is more efficient than MSC-TERT cells in other genes causing bone monogenic disorders
Next, we addressed the possibility of the FGFR2 locus being a prime editing-resistant locus in the genome of MSC-TERT cells. Given the evolutionary conservation of the gene and its central role in diverse cellular processes,8,36–38 it is plausible that FGFR2 is exceptionally protected against ss nucleotide substitutions.
First, to zoom in on the FGFR2 gene and assess whether it is specifically the p.C342 site that is resistant, we attempted to introduce another prevalent syndromic craniosynostosis mutation: FGFR2-S252W (c.755 C > G; p.S252W; Apert syndrome). We transfected both cell lines with a PE5 −36 ngRNA setup and observed an editing efficiency of 0.2% for the HEK293FT cells by NGS analysis. No edits were detected for MSC-TERT cells (Table 1).
Overview of prime editing attempts in various other bone disease related genes
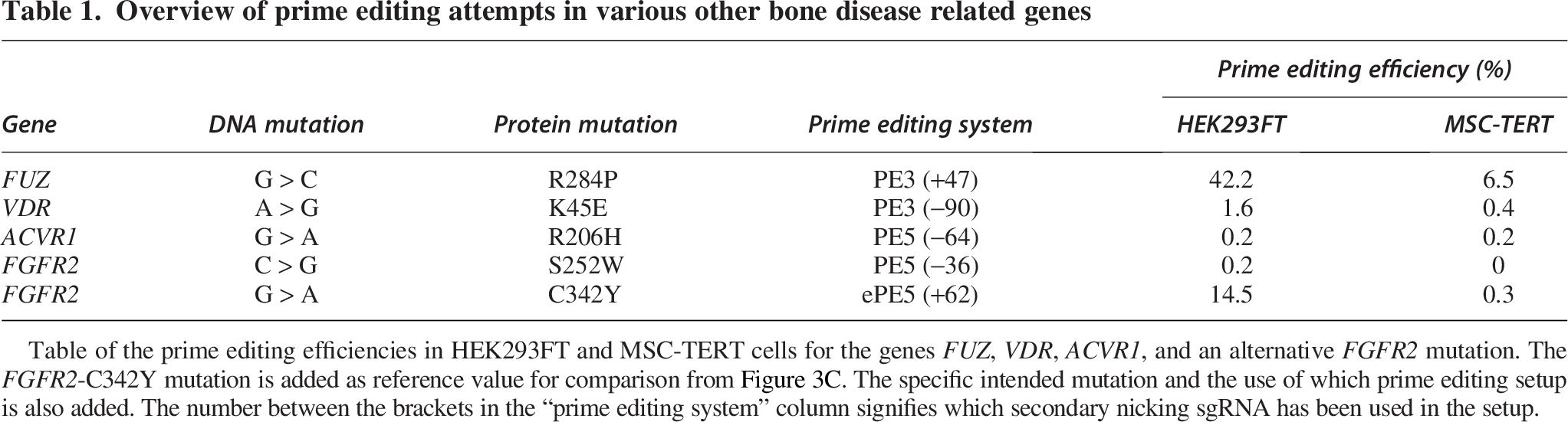
Table of the prime editing efficiencies in HEK293FT and MSC-TERT cells for the genes FUZ, VDR, ACVR1, and an alternative FGFR2 mutation. The FGFR2-C342Y mutation is added as reference value for comparison from Figure 3C. The specific intended mutation and the use of which prime editing setup is also added. The number between the brackets in the “prime editing system” column signifies which secondary nicking sgRNA has been used in the setup.
Next, we targeted genes involved in bone monogenic disorders other than FGFR2: FUZ-R284P (craniosynostosis), vitamin D receptor (VDR)-K45E (rickets), and activin A receptor type 1 (ACVR1)-R206H (fibrodysplasia ossificans progressive), and analyzed them by NGS.35,39,40 When introducing the FUZ-R284P, we observed a higher editing efficiency in the HEK293FT cells compared with the MSC-TERT cells (Table 1, Supplementary Fig. S3). When introducing the VDR-K45E, we observed an editing efficiency of 1.6% in HEK293FT cells but did not observe any editing in MSC-TERT cells. Prime editing attempts for ACVR1-R206H yielded no detectable edits in either cell line. Comparing the editing efficiencies of all target genes in MSC-TERT cells, we observed an editing efficiency above 0.5% in MSC-TERT cells only when introducing the FUZ-R284P mutation (Table 1). This supports that MSC-TERT cells could be overall less susceptible to prime editing, but can still be edited depending on the locus targeted.
Introduction of a synonymous mutation in FGFR2 does not increase the success rate of prime editing in MSC-TERT cells at FGFR2
Although MSC-TERT cells demonstrated susceptibility to prime editing, as evidenced by the successful incorporation of the FUZ-R284P and HEK3-CTTins mutations, we did not detect any successfully incorporated FGFR2-C342Y mutations in the MSC-TERT cells with an optimized prime editing system. We considered the possibility that introducing FGFR2 mutations may be detrimental to MSC-TERT viability. To investigate this, we transfected the cells with a new pegRNA that targets the same PAM site as the FGFR2(C342Y) + 62 pegRNA, but instead introduces a synonymous substitution (c.1027_1029delinsCTC) in the Lysine codon on position 343 (hereafter termed “FGFR2(L343L) + 62”). This approach enables detection of genomic edits without altering the amino acid at position p.Cys342, preserving the native FGFR2 protein (Fig. 3B).
Transfection of FGFR2(L343L) + 62 constructs into HEK293FT successfully introduced its mutation with slightly higher efficiencies as FGFR2(C342Y) + 62 in PE3 (2.7% for L343L; 1.8% for C342Y) and PE5 (8.9% for L343L; 5.4% for C342Y) setups, based on the restriction enzyme digestion analysis and NGS analysis (Fig. 3B). Similar to the results from the FGFR2(C342Y) + 62 prime editing, we again observed a lower editing efficiency when using the PEmax-Cas9 protein (PE5max: 7.9%) compared with the use of PE-Cas9 (PE5: 8.9%) (Fig. 3B). No successful edits were observed in the MSC-TERT conditions. This observation shows that the absence of successfully incorporated craniosynostosis mutations in MSC-TERT FGFR2 is not due to the generation of a nonfunctional FGFR2. Furthermore, it supports the rationale that the FGFR2 locus is difficult to edit with prime editing in MSC-TERT cells.
The Crouzon C342Y mutation is detected in the nonadherent (floating) cell fraction of MSC-TERT cultures
MSC-TERT and HEK293FT cells are primarily cultured attached to a surface. However, we also detected a fraction of detached, floating cells. We investigated to what extent we can detect the FGFR2-C342Y edit in this nonadherent FCF of the MSC-TERT cultures. For this purpose, we transfected MSC-TERT cells with the ePE5 FGFR2(C342Y) + 62 set-up. We collected the FCF after 72 h (t = 3 days) and 168 h (t = 7 days) when culture medium was refreshed. In addition, we also collected the adherent cell culture at day 7.
We observed successful incorporation of the FGFR2-C342Y mutation together with the PAM-site ablating synonymous mutation in the FCF of the MSC-TERT cells at day 3 (Fig. 4A). The incorporation was observed in the FCF at day 7 as well, albeit less prevalent compared with day 3 based on the height of the A-nucleotide peaks (Fig. 4A). In line with the previous observations, we do not detect any mutation in the attached cells of the same culture at day 7.
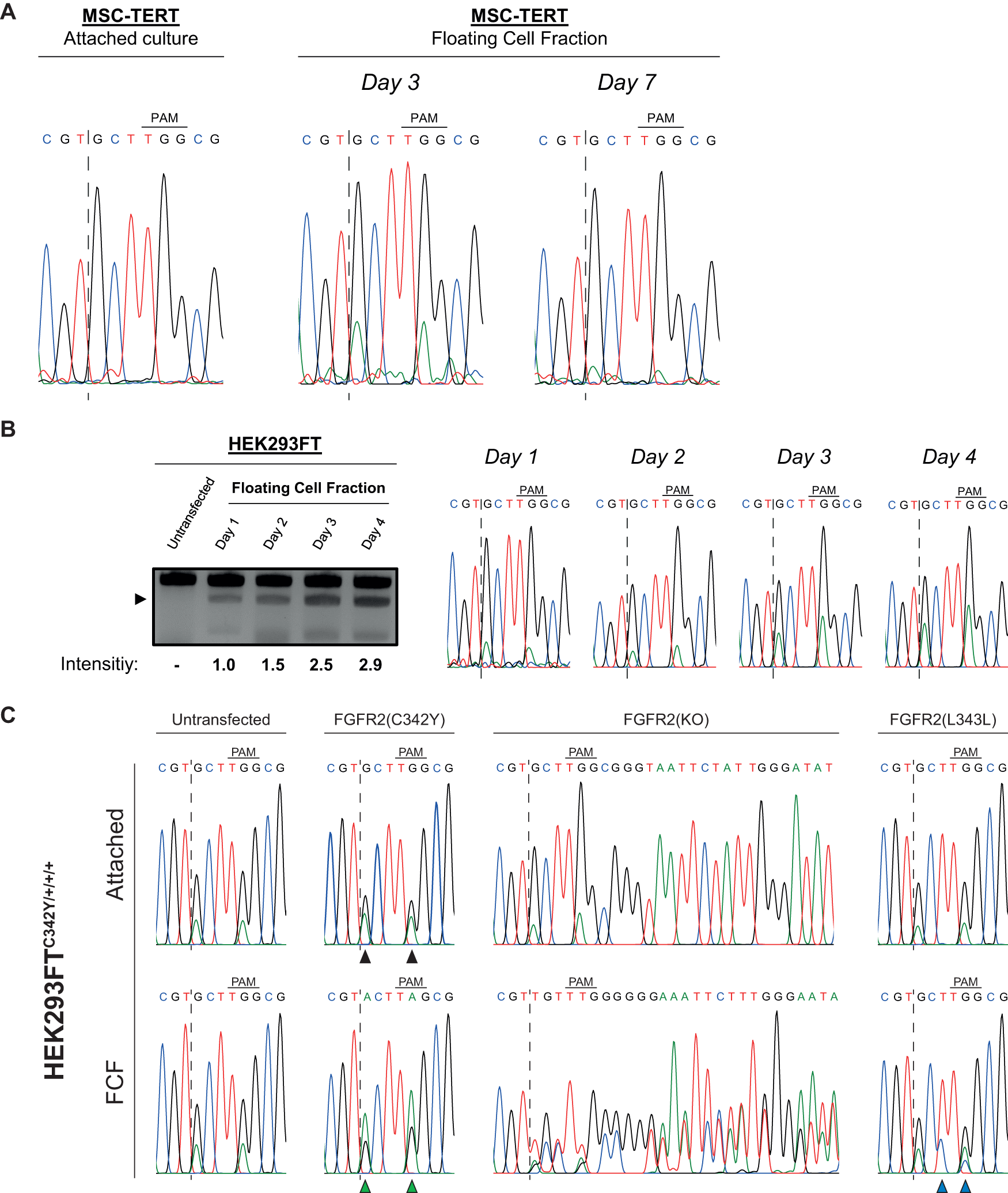
Analysis of the floating cell fraction (FCF) in MSC-TERT, HEK293FT, and HEK293FTC342Y cells after transfection of prime editing constructs.
It is currently unclear whether the MSC-TERT cells detach because of the introduced mutation or if editing and incorporation of the mutation takes place when the cells are already detached and floating. To test this latter possibility, we transfected HEK293FT cells with the ePE5 FGFR2(C342Y) + 62 setup since HEK293FT cells have already demonstrated to yield a relatively high percentage of edits with this prime editing set-up in attached cells (Fig. 3C). In this way, we ensure that a functioning prime editing setup is being tested when assessing editing in the floating cells. At 24 h post-transfection, we collected the culture medium containing the FCF and divided the fraction into four separate wells. We cultured these cell fractions, collected gDNA of one of the wells every 24 h, and analyzed for the presence of our FGFR2-C342Y edit by RsaI enzymatic digestion in the respective PCR product.
We detected RsaI digested PCR product in the FCF of HEK293FT cells 24 h after transfection (t = 1 day), which increased by almost threefold over the following 4 days (Fig. 4B). These data confirm the presence of the FGFR2-C342Y mutation in the FCF and provide a reason that editing continues for at least 4 days while the cells are floating in culture.
Editing of HEK293FTC342Y/+/+/+ cells exhibits higher editing efficiencies in floating cells compared with their attached counterpart
Next, we want to address the possibility of the incorporated FGFR2-C342Y mutation triggering a cellular response leading to detachment from the surface. As described above, the FGFR2-C342Y mutation is found in the FCF of MSC-TERT cells rather than in the attached fraction of the culture. This made us question whether the detachment of the cells is a consequence of incorporating the mutation in half or more of the FGFR2 alleles. Since our HEK293FTC342Y/+/+/+ cell line is tetraploid for FGFR2, we can use this cell line as a tool to investigate this hypothesis by systematically targeting the remaining three wild-type alleles and introduce either the FGFR2-C342Y mutation again, the synonymous FGFR2-L343L mutation, or knocking out the whole allele.
To this end, HEK293FTC342Y/+/+/+ clones 1 and 2 were transfected with either the ePE5 FGFR2(C342Y) + 62 constructs, the ePE5 FGFR2(L343L) + 62 constructs, or a conventional CRISPR-Cas9 knockout targeting of FGFR2 by introducing DSBs and the subsequent nonhomologous end joining (NHEJ) repair resulting in indels.
When transfecting both HEK293FTC342Y/+/+/+ clones with the ePE5 FGFR2(C342Y) + 62 constructs, we found that the presence of the FGFR2-C342Y mutation was increased in the attached cultures, compared with the untransfected cells. However, the presence of the FGFR2-C342Y mutation was increased even more in the FCF of both clones, exceeding the presence of the WT copies (Fig. 4C; Supplementary Fig. S6). We found no traces of successful incorporation of the FGFR2-L343L mutation in the attached cultures of neither clone 1 nor 2. The FGFR2-L343L mutations were successfully incorporated, however, into the genome in the FCF of both clones (Fig. 4C; Supplementary Fig. S6). Transfecting the HEK293FTC342Y/+/+/+ clones with conventional CRISPR knockout constructs resulted in a drastically higher amount of +1 insertions in the FCF (clone 1—49.7%; clone 2—39%) compared with the attached fractions (clone 1—no indels; clone 2—6.8%) (Fig. 4C; Supplementary Figs. S6 and S7).
Discussion
This study explored the possibilities of utilizing prime editing to generate an in vitro disease model for syndromic craniosynostosis research. Our objective was to introduce a high-prevalence craniosynostosis-associated FGFR2 mutation (c.1025 G > A; p.C342Y) into both HEK293FT cells as well as the immortalized osteoprogenitor MSC-TERT cell lines. The in vitro disease models generated through this approach would enable efficient modeling of the diverse mutations associated with craniosynostosis in a consistent and stable cell line within a relatively short timeframe compared with in vivo approaches. Within this study, we confirmed that prime editing is a feasible gene-editing tool in both HEK293FT and MSC-TERT cells, albeit with varying efficiency.
We observed variation in prime editing efficiencies between the MSC-TERT and the HEK293FT cells at various genomic target sites. Introducing the FGFR2-C342Y, HEK3-CTTins, FUZ-R284P, and VDR-K45E mutations was more efficient in HEK293FT cells compared with MSC-TERT cells. When directly comparing editing efficiencies between the FGFR2, HEK3, FUZ, and VDR loci within the same cell type, we also find a high degree of variation in prime editing efficiency between the loci. These observations are in agreement with other studies investigating factors determining prime editing efficiencies, which point toward both a cell-based variance as well as a locus-based variance.27,29,30,34,41–43 However, when introducing FGFR2-S252W and ACVR1-R206H, both mutations were incorporated with an editing efficiency below 0.3% in both cell types. Since editing of the HEK293FT cells was also unsuccessful, this may point either at dysfunctional prime editing constructs or at inaccessible target sites.
In accordance with published literature, we observed an increased prime editing efficiency when adding a secondary ngRNA (PE3) over the PE2 system when targeting FGFR2-C342Y and FUZ-R284P. Similarly, we also observed variance in prime editing efficiency between applied secondary ngRNAs within the same locus.27,29,44,45 These data and their accordance with literature underline the challenge of prime editing when selecting suitable secondary ngRNAs and the laborious effort to screen for an effective nicking guide.
The absence or very low editing in MSC-TERT cells at the FGFR2 locus led us to investigate several previously reported optimization approaches to enhance the prime editing efficiency. In some cases, these optimizations did lead to editing in cell types that were initially not susceptible to prime editing attempts due to mismatch repair activity.29,30 Despite these optimization attempts, we did not observe any improvement or successfully incorporated mutations in MSC-TERT cells. In contrast, prime editing efficiency was noticeably increased in HEK293FT cells in all conditions where the optimization constructs were included. These findings suggest that the applied optimization strategies can enhance the prime editing efficiency at this locus in HEK293FT cells, but not in MSC-TERT cells. The reason for the different performance at the FGFR2 locus in both cell types remains elusive.
Closed chromatin states impede editing constructs from binding or interacting with their genomic targets.46–51 In addition, altering the epigenetic landscape of the genome can have beneficial effects on gene-editing efficiencies.52–54 With this in mind, we assessed whether genome-editing machinery can reach and interact with its FGFR2 target in MSC-TERT cells. When we transfected MSC-TERT that stably expresses endogenous spCas9, with the FGFR2(C342Y) pegRNA, we observed indels as a result of NHEJ. Hence, although no edits are introduced with prime editing, DSBs and NHEJ still occur at the FGFR2 locus when utilizing a conventional Cas9 nuclease and a pegRNA. This makes it unlikely that the FGFR2 locus of MSC-TERT cells is excessively compacted within the chromatin and does not explain the low editing efficiency compared with the same locus in HEK293FT cells.
A large question within our study remains whether the elevated prime editing in the FCF of HEK293FT and MSC-TERT cells is a cause or a consequence of cell detachment. Simultaneously, we questioned whether these detached cells are still viable or whether apoptosis has occurred. Disruption of cellular FGFR2 homeostasis by introduction of the C342Y mutation could affect cell survival in diploid genomes. Tyrosine kinase activity of FGFR2 is mediated by the dimerization of FGFR2 monomers. Various mutations in FGFR2 disrupt the disulfide bonding between cysteine residues of FGFR2 monomers, allowing or preventing spontaneous dimerization. Ablation of both cysteine residues p.Cys278 and p.Cys342 in the IgIII loop responsible for disulfide binding results in the inability to promote tyrosine kinase activity. 55 In vivo, a homozygous FGFR2-C342Y mutation in mice is unfit for fetal survival as pups die within a short time after birth.56–58 Similarly, mice with a homozygous disruption of the IgIIIc domain, as well as mice hemizygous for this domain, die within a short timeframe postnatally, stressing the significance of a functional IgIIIc domain.59–61 A major genomic difference between our two cell lines is the tetraploidy we observed for FGFR2 in HEK293FT cells,62,63 whereas the MSC-TERT cells are diploid. 26 When only two copies of FGFR2 are present, of which one is mutated, its incorporation may lead to dysregulation of FGFR2 signaling, potentially triggering apoptosis in the affected cells. In contrast, the additional three healthy FGFR2 copies within our HEK293FTC342Y/+/+/+ cells could buffer the imbalance caused by one mutated allele, maintaining the viability of the cells with the incorporated mutation. However, we do not possess sufficient data to credibly conclude whether these cells are indeed going into apoptosis after incorporation of the FGFR2-C342Y mutation.
Alternatively, the incorporation of the FGFR2-C342Y mutation could negatively affect cell-matrix interactions, resulting in detachment from the culture surface rather than going into apoptosis. FGFR2 is reported to interact with various cell adhesion molecules (CAMs), such as NCAMs, FLRTs, and IgLONS. 64 To our knowledge, no thorough studies have been published on the effect of the Crouzon C342Y mutation in FGFR2 on its interactions with CAMs. However, disruptions in FGFR2 signaling have been shown to affect the functionality of CAMs in several other studies. Silencing FGFR2 resulted in reduced cell adhesion in human mammary epithelial (HB2) cells. 65 Loss of Fgfr2 signaling in mice and immortalized frontonasal prominence cells affect cell–matrix adhesion. Cell adhesion was defective in cells where FGFR2 was absolved of its kinase activity (kinase dead).61,66 Apert syndrome is the result of a p.S252W substitution mutation in FGFR2. It does also lead to receptor gain of function, although the receptor is not constitutively activated as in Crouzon C342Y. In Apert S252W affected cells, several cell adhesion genes are downregulated, the cells show lower individual spreading of the cell membrane, and cells show a decreased cell attachment when cultured in vitro on collagen or fibronectin.67,68 Despite the lack of publications on the impact of Crouzon C342Y on the interaction between FGFR2 and CAMs, cell adhesion is affected and altered upon loss of FGFR2 signaling and in the presence of a similar FGFR2-linked craniosynostosis syndrome. Both C342Y and S252W are located on (C342Y) or near (S252W) the IgIII domain of FGFR2, and FGFRs are likely to interact with NCAM1 through that IgIII domain. 69 Therefore, we speculate a possibility that the Crouzon FGFR2-C342Y mutation we introduce in this study has an effect on cell adhesion, thus explaining why we observe a superior presence of mutation in the FCF. Although MSC-TERT cells do not appear to express NCAM1, 70 osteoblasts from fetal coronal sutures do express NCAM1 and could therefore be a relevant CAM to investigate further how its interaction with FGFR2 is altered by the Crouzon C342Y mutation. 71
Surprisingly, the introduction of the FGFR2-C342Y mutation in HEK293FTC342Y/+/+/+ cells resulted in a higher presence of the mutation in detached cells compared with the respective attached cells. Similarly, the introduction of random indels which disrupt the healthy alleles was most observed in the FCF. However, when introducing the synonymous FGFR2-L343L (c.1027_1029delinsCTC) mutation, we again observe an increased editing frequency in the FCF, but not in the attached cells. This observation suggests that it is more likely that this nondisruptive mutation is introduced after cells have already detached from the surface. This is supported by our data on the continuous incubation of the HEK293FT FCF over 4 days, in which the presence of the FGFR2-C342Y mutation progressively increases over time despite the lack of any attached cells. With this, we speculate that only once the MSC-TERT cells detach, that the cells become susceptible for the editing machinery on FGFR2 by circumventing an unknown safeguarding mechanism active in attached cells.
Conclusions
The successful generation of the HEK293FTC342Y/+/+/+ cell line demonstrates the possibility of establishing a human cell line with specific craniosynostosis mutations, enabling the investigation of the molecular and functional consequences of these mutations without relying on transgenic overexpression approaches. The MSC-TERT cells that were proposed to fulfill this same solution tend to be less susceptible to prime editing of FGFR2, HEK3, FUZ, and VDR compared with the HEK293FT cell line. Nevertheless, we do point out that the MSC-TERT cells are not resistant to prime editing, as we have shown successfully introduced mutations in HEK3 and FUZ. The successful incorporation of these mutations provides confidence in the MSC-TERT cells as a suitable in vitro disease model, even though the success may depend on the targeted locus. This is supported by the serendipitous finding that the FGFR2-C342Y mutation in the MSC-TERT cells is found in cells that are not attached to a surface, but rather part of the FCF. Similarly, the synonymous FGFR2-L343L mutation was found back in the FCF of HEK293FTC342Y/+/+/+ cells, rather than the attached cells. These observations are also of general interest for the application of prime editing as it implies that its editing could be more efficient in cells when they are not attached to a surface. Although a concrete explanation for this observation remains elusive, it is suggestive that cytoskeletal and nuclear matrix organization, as well as cellular morphology, might contribute to the success rate of prime editing.
Authors’ Contributions
M.G.: Formal analysis (lead), investigation (lead), methodology (lead), project administration (supporting), resources (supporting), validation (equal), visualization (lead), and writing—original draft (lead). F.M.D.: Investigation (supporting), validation (supporting), and visualization (supporting). A.F.M.: Formal analysis (supporting), investigation (supporting), and visualization (supporting). T.M.: Formal analysis (supporting) and investigation (supporting). M.K.: Resources (equal). J.v.d.O.: Conceptualization (supporting), supervision (supporting), validation (supporting), and writing—review and editing (supporting). I.M.J.M.: Conceptualization (equal), funding acquisition (equal), and writing—review and editing (supporting). J.P.T.M.v.L.: Conceptualization (lead), funding acquisition (lead), project administration (equal), supervision (equal), and writing—review and editing (equal). J.v.d.P.: Conceptualization (equal), data curation (lead), funding acquisition (equal), methodology (supporting), project administration (lead), resources (lead), software (lead), supervision (lead), validation (lead), and writing—review and editing (lead).
Footnotes
Author Disclosure Statement
No competing interest declared.
Funding Information
This work was supported by the Erasmus MC Grants (
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.