
Abstract
The endoscopic Functional Lumen Imaging Probe (FLIP) devices are used to evaluate pressure changes, diameter, and volume of the esophagus. We used the FDA’s MAUDE database to collect post-marketing surveillance data on these devices from January 2009 to September 2022. Forty-Five device-related events and thirty-six patient-related adverse events were analyzed. The most common device issue for the diagnostic FLIP device was therapeutic/diagnostic failure (n = 6), while the most frequent issue with the therapeutic FLIP device was adverse events without an identified device or use problem (n = 11). Patient-related adverse events were extremely rare with the diagnostic FLIP and the most common patient-related adverse event with the therapeutic FLIP was perforation (n = 11). Endoscopists need to be mindful of these potential technical issues and adverse events while using these devices.
Introduction
Esophageal motility is an integral part of esophageal disorders, and motility testing is essential for diagnosis. 1 Over time, techniques for measuring esophageal and sphincter physiology have evolved. High-resolution manometry using esophageal pressure topography is the current standard, 2 and bolus transit is now described more comprehensively with simultaneous impedance analysis. 3 The introduction of endoscopic functional luminal imaging probes (FLIP) has allowed for the measurement of the mechanical properties of the esophagus in comparison to contraction patterns and bolus transit. 4
The FLIP devices, EsoFLIP® (Medtronic Inc, Shoreview, MN, USA) and EndoFLIP® (Medtronic Inc, Minneapolis, MN, USA), use impedance planimetry during volume-controlled distention to estimate the luminal cross-section area (CSA) of the esophageal lumen. 5 The FLIP concept was first demonstrated in 2006, and the diagnostic FLIP device has been commercially available since 2009, and shortly after that, the therapeutic FLIP was introduced. 6 These disposable devices provide real-time visualization of the esophagus.7,8 Diagnostic FLIP (EndoFLIP®), is used to measure pressure and cross-sectional area, while the therapeutic FLIP (EsoFLIP®) was designed for esophageal dilation.9,10 The FDA has approved diagnostic FLIP for measuring esophageal, pyloric, and anal sphincter dimensions and pressures, estimating gastric band stoma size, and for use as an adjunctive test in esophageal hypersensitivity. 10 The American Gastroenterological Association Institute’s expert review provided essential guidance on using FLIP in clinical practice, such as using it as a complementary tool to assess opening dynamics of the esophagogastric junction and esophageal wall stiffness and utilizing it for severity assessment of eosinophilic esophagitis and therapeutic monitoring. 5
While studies have demonstrated the effectiveness of FLIP devices in these procedures, information regarding associated adverse events is limited. This study aims to evaluate adverse events and complications associated with using FLIP devices by analyzing the data available in the FDA’s Manufacturer and User Facility Device Experience (MAUDE).
Methods
We analyzed post-marketing surveillance data for diagnostic FLIP (EndoFLIP®) and therapeutic FLIP (EsoFLIP®) devices using the FDA’s MAUDE database to report patient-related adverse events and device-related issues. The data are accessible at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm. The MAUDE database collects reports related to medical devices, including suspected device-related deaths, serious injuries, and malfunctions that the FDA uses to monitor the devices after FDA approval. Medical device reports may be submitted by mandatory and voluntary reporters, including healthcare professionals, patients, and consumers.
In this study, we analyzed medical device reports from the MAUDE database between January 2009 and September 2022. Search terms included EsoFlip or EndoFLIP under the brand name. We individually reviewed each report for event date, device malfunction type, device-related problems, patient-related adverse events, and mortality. Each report can have one or more device or patient-related issues. Statistical analysis was performed using Microsoft Excel 2010 (Microsoft Corporation, Redmond, Wash, USA). The results are presented as percentages for categorical variables and mean standard deviation or medians with interquartile range (IQR) for quantitative variables. No IRB approval was needed as the data was obtained from a publicly available de-identified database.
Results
From January 2009 to September 2022, 45 device-related events and 36 patient-related adverse events were submitted. The first report was submitted to the MAUDE database in 2017, with zero reports/events from 2009 to 2016. The reason for no reports from 2009 to 2016 is likely the limited adoption of the FLIP devices in the initial years; however, the exact reasons cannot be elucidated.
Device-Related Problems
Forty-five device events were reported, including 29 related to the diagnostic FLIP and 16 related to the therapeutic FLIP device.
Device-related problems associated with the diagnostic FLIP device
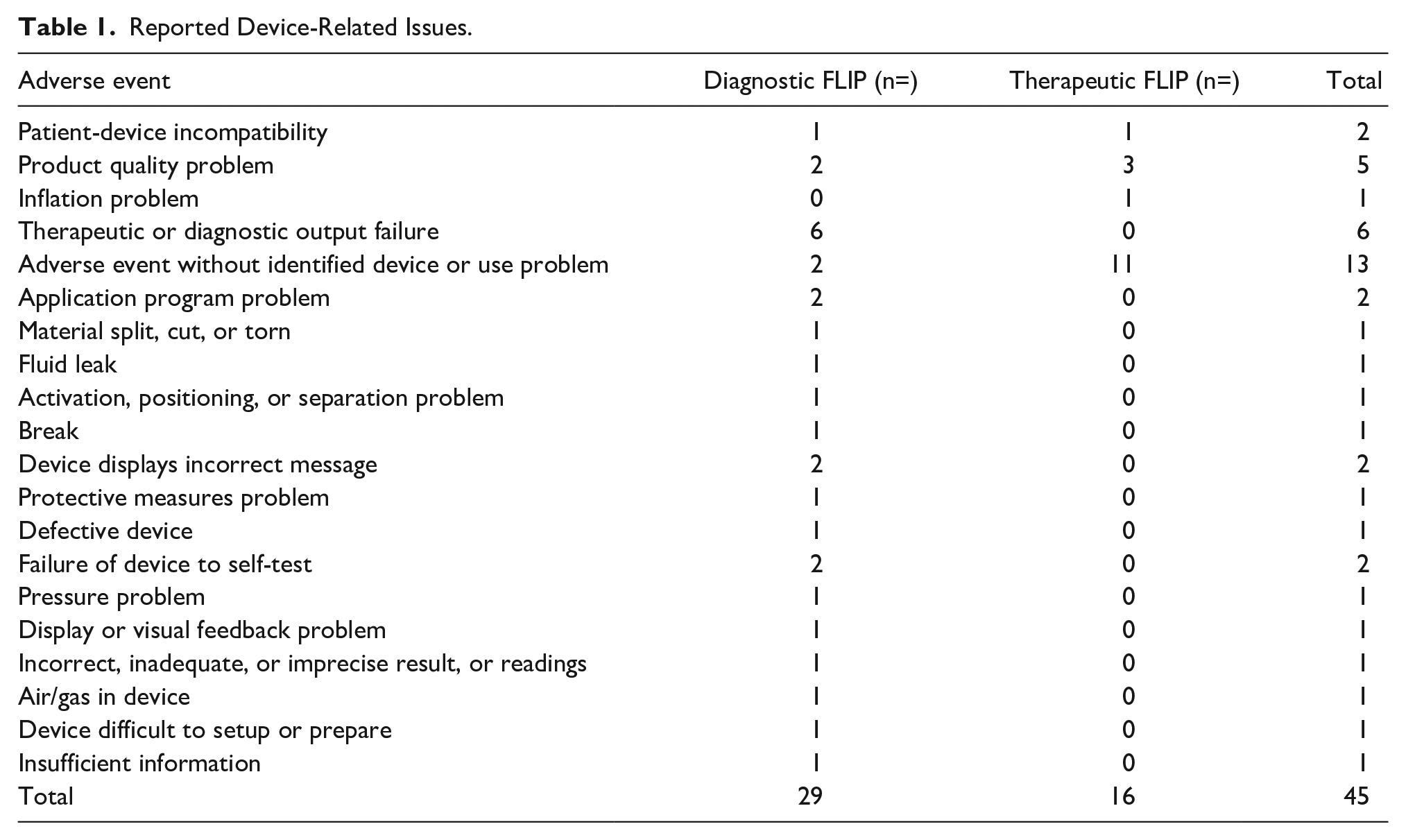
The most common diagnostic FLIP device issues were therapeutic/diagnostic failure (n = 6), product quality problems (n = 2), adverse events without an identified device/use problem (n = 2), the device displaying an incorrect message (n = 2), failure of the device to self-test (n = 2) and application program problem (n = 2). Other device problems are listed in Table 1. Product quality issues involved catheter misconnection and improper syringe locking, but no patient harm was reported.
Reported Device-Related Issues.
Device-related problems associated with the therapeutic FLIP device
For the therapeutic FLIP device-related issues, adverse events without an identified device or use problem were the most commonly reported issue, with 11 events, followed by product quality problems, which accounted for 3 events. The reported product quality problems included catheter failure at the start of the procedure, improper loading of the syringe leading to temperature sensor failure error requiring repeat procedure and additional anesthesia, frozen system screen, and inability to report if the catheter continued to inflate. There were no reports of patient or user harm. Other device-related problems included patient-device incompatibility and inflation problems, with 1 event each.
Patient-Related Adverse Events
Thirty-seven patient-related adverse events were reported in the MAUDE database over the study period, consisting of 36 events listed in Table 2. Most were submitted in 2019 (n = 22), followed by 6 reports in 2021 and 2022. Most event types were malfunctions (n = 22, 59.5%), followed by injury (n = 14, 37.8%) and death (n = 1, 2.7%).
Reported Adverse Patient Events.
Patient-related adverse events associated with diagnostic FLIP
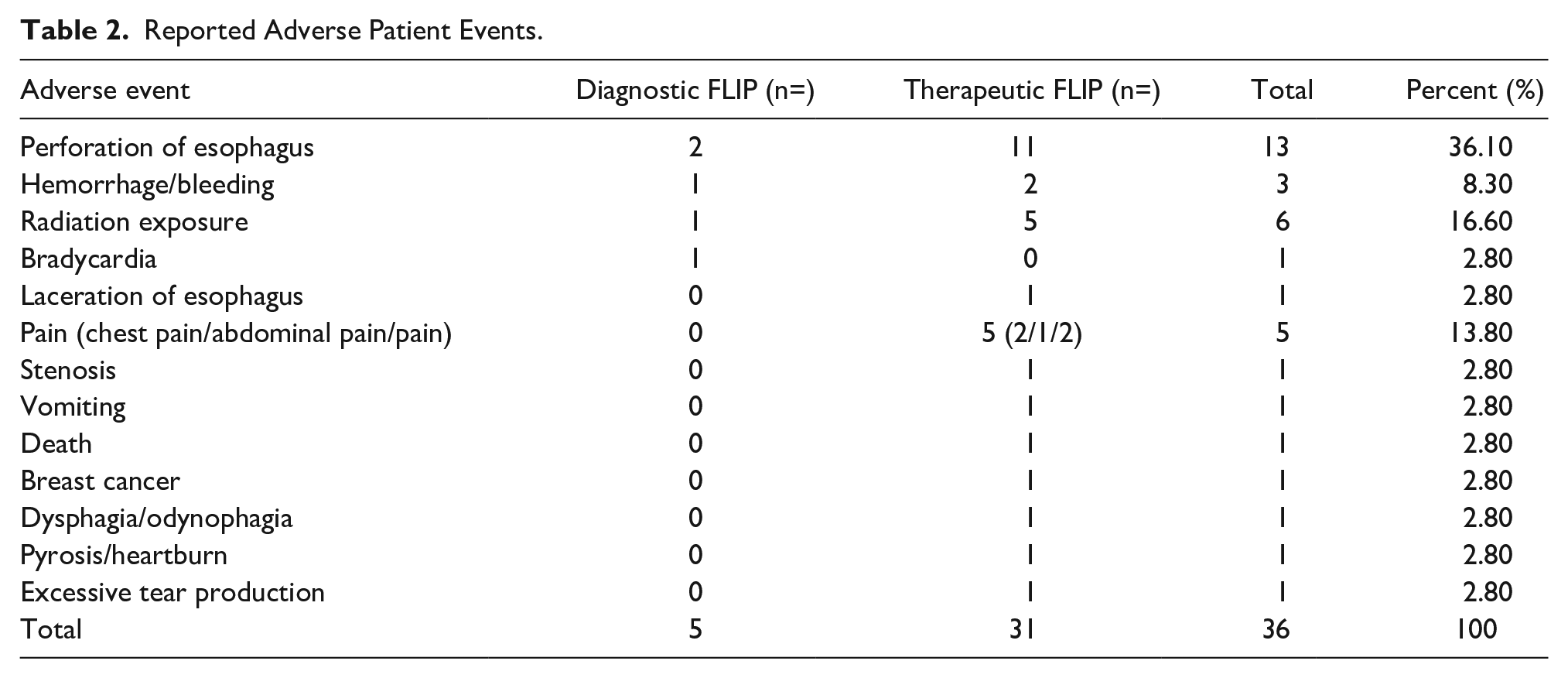
The most common patient-related adverse event among diagnostic FLIP was esophageal perforation (n = 2), followed by radiation exposure (secondary to imaging study), pain, and bleeding/hemorrhage, with one each.
Patient-related adverse events associated with therapeutic FLIP
The most common patient-related adverse event among therapeutic FLIP was esophageal perforation (n = 11), followed by radiation exposure (secondary to imaging study) (n = 5), pain (n = 5), and bleeding/hemorrhage (n = 2). Other adverse events are listed in Table 2.
Esophageal Perforation
Esophageal perforation was the most frequently reported adverse reaction, with the majority occurring with the use of the therapeutic FLIP (n = 11). The 2 reported perforations with the use of diagnostic FLIP were in conjunction with pneumatic dilation and blind insertion of the catheter during the POEM procedure. Therefore, none of these events can be independently attributed to the appropriate use of the FLIP device itself.
One death was reported in a patient who underwent dilatation using therapeutic FLIP for type 2 achalasia. The patient had metastatic breast cancer, which caused a stricture in the esophagus. However, the information provided in the claim is not enough to suggest that the death can be attributed to the use of the FLIP device. Twelve patients survived the perforation events, with different management methods required.
Discussion
This study analyzed adverse event reports associated with diagnostic and therapeutic FLIP devices from the MAUDE database from January 2009 to September 2022. Forty-five device-related events and 36 patient-related adverse events were reported during this period. The most common device issue was therapeutic/diagnostic failure (n = 6) for diagnostic FLIP, while adverse events without an identified device or use problem (n = 11) were the most frequently reported issue with the therapeutic FLIP. Patient-related adverse events were extremely rare with the diagnostic FLIP and the most common patient-related adverse event with the therapeutic FLIP was perforation.
The incidence of esophageal perforation varies between different reports and modalities of esophageal dilatation. Analysis of 29 studies evaluating pneumatic dilatation in achalasia found the perforation rate to be 2% (24/1358). 11 Another single-center retrospective study evaluating pneumatic dilatation in achalasia analyzed 830 pneumatic dilation procedures performed on 372 patients, and found 16 reports of transmural esophageal perforation (4.3% of patients, 1.9% of dilations). 12 A meta-analysis of 5 RCTs comparing bougie and endoscopic balloon dilatation in patients with benign esophageal strictures found an overall incidence of esophageal perforation of 1.05% and 0.37%, respectively, with no between-group risk difference of 0.01 (95% CI [0.03; 0.02]). 13 Studies evaluating esophageal perforation rates after therapeutic FLIP are scarce. A retrospective study of 17 patients undergoing therapeutic FLIP-guided dilatation for achalasia identified 1 case of esophageal perforation requiring surgical intervention. 14 Further studies comparing different esophageal dilation modalities are needed.
Our analysis found that the most common reported device issue was therapeutic/diagnostic failure (n = 6) for diagnostic FLIP, followed by product quality problem, application program problem, failure of the device to self-test, and display of the incorrect message, with 2 each. For the therapeutic FLIP, adverse events without an identified device or use problem (n = 11) were the most frequently reported issue. Reports did not provide specific information about why each device-related adverse event occurred, but other reports mentioned technical difficulties such as computer freezing, balloon inability to inflate, and inaccurate pressure and temperature readings. Most device-related adverse events were due to computer problems and catheter malfunctions. Additional studies are needed to examine the underlying causes of these adverse events and device failures.
The most common patient-related adverse events were esophageal perforations. These perforations required surgical intervention on 5 occasions, conservative management on 3 occasions, stent placement on 3 occasions, death of 1 patient, and undocumented treatment in 1 report. The single reported event of death with the use of the therapeutic FLIP device for esophageal dilatation cannot be clearly attributed to the FLIP device, given the underlying malignancy in this patient, as mentioned above. Two cases of perforations with the diagnostic FLIP have been reported. The first case was reported as occurring following a pneumatic dilation, but it implies that it was actually from therapeutic dilation and, thereby, is likely misreported as using the diagnostic FLIP. The second case of perforation reported with the diagnostic FLIP occurred during blind placement of the FLIP catheter after creation of the submucosal tunnel during the POEM procedure, which is not the standard procedure for the use of this device. According to expert consensus by Su et al, 15 blind placement of the FLIP catheter in conjunction with a POEM procedure can result in esophageal perforation, and thus placement should always be made under direct endoscopic visualization. Therefore, neither of the reported perforations with the diagnostic FLIP can be independently attributed to the appropriate use of the device itself. This also highlights the potential limitation of the MAUDE database since reporting of adverse events is not standardized and can be subject to inadvertent misreporting.
Our study has several limitations, most are due to the inherent nature of the MAUDE database. The database lacks consistency and detail in reporting, with submissions made by various healthcare professionals who may not have direct involvement in the adverse events or patients themselves. This can result in incomplete, inaccurate, or insufficient entries that may miss important data points. Adverse events may therefore be underreported or sometimes unrelated to the FLIP devices. There are also duplicate entries in the database that can skew data, and procedural details may be missing, making it challenging to determine the exact causes or consequences of events. The adverse events are reported as numbers without denominators due to the lack of the total number of devices over that period, making it difficult to calculate the frequency of these adverse events. Lastly, when using FLIP devices, it can be challenging to distinguish association from causality for certain reported adverse events, such as esophageal perforation, which may be due to the nature of the disease rather than the device use itself.
Despite these limitations, our analysis provides information regarding the potential adverse events associated with using FLIP devices. Our analysis also demonstrates that the diagnostic FLIP device is safe in routine clinical practice.
Footnotes
Acknowledgements
None.
Author Contributions
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.