
Abstract
Background:
Acinetobacter baumannii is a common bacterial pathogen in nosocomial infections. It has become one of the greatest threats to human health for its growing resistance to “last resort” antibiotics, which has led to a revival of phage therapy as a potential treatment. However, conventional methods for isolating A. baumannii-infecting phages are labor-intensive and often unsuccessful.
Methods:
Our approach involves a computational pipeline to identify temperate phages (prophages) integrated into A. baumannii genomes, followed by mitomycin C (MMC) induction of those strains to screen for active prophages.
Results:
Here we show a prophage analysis for nearly 900 A. baumannii genomes. We observed MMC-triggered excision of nine prophages from eight A. baumannii strains by Polymerase Chain Reaction (PCR) and sequencing. Further, we show four prophage-formed virions detectable by transmission electron microscopy and two that can plaque on other A. baumannii isolates.
Conclusion:
This work demonstrates the utility and diversity of prophages for further development as therapeutics for antibiotic-resistant A. baumannii.
Introduction
Antibiotic resistance (AR) is a significant threat to global public health; 4.95 million deaths across the world were estimated to be associated with bacterial AR in 2019, 3 highlighting the need for novel and potentially diverse therapeutics against antibiotic-resistant pathogens. The six “ESKAPE” organisms (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter sp.) are of particular interest due to their alarming rise in AR and high rates of infection. Carbapenem-resistant A. baumannii in particular is ranked as a critical-priority pathogen by the World Health Organization based on factors such as limited treatability, prevalence, and infection mortality.43,54 In some countries, rates of carbapenem resistance in A. baumannii can be as high as 80–90%. 36
Bacteriophages (phages) are the natural predators of bacteria that have been studied extensively for longer than a century. 1 AR has led to a revival of leveraging phages as therapeutic agents against infections (“phage therapy”5,21). Across the world multiple patients have been treated using phage therapy for different antibiotic-resistant bacterial pathogens.1,28,45,50 Despite these successes, phage therapy faces significant implementation challenges. While lytic phages isolated from environmental samples are the gold standard for phage therapy applications, reliance solely on environmental isolation may not provide sufficient diversity for large phage banks, or phage-derived enzyme therapies such as lysins may not be fulfilled with only lytic phages.19,29 Further, phages often exhibit narrow host ranges with strain-level specificity, making it challenging and time intensive to find phages for specific target strains.
One approach to increasing the diversity of available phages for antibiotic-resistant pathogens is to explore the use of prophages. Prophages are a class of chromosomal mobile genetic elements that have integrase proteins, which allow them to integrate their genomes at specific sites in the bacterial chromosome. They are typically overlooked as therapeutics due to concerns about transduction, lysogenic conversion, and the potential to increase virulence through horizontal gene transfer.8,38,46 However, prophages have been converted into virulent phage through genome engineering that removes integration machinery and other potentially deleterious gene products.10,22,33 This approach provides researchers with an alternate method to access a large array of phages when traditional phage discovery is unsuccessful.
In this study, we precisely identified prophages within the genomes of a panel of 891 antibiotic-resistant A. baumannii strains using Targeted Integrative Genetic Element Retriever (TIGER) and Islander software 33 and identified 789 precisely predicted prophages. Comparison of this dataset to previously isolated A. baumannii phages deposited in GenBank revealed increased diversity, suggesting that the prophages provide an expanded pool of phages for therapeutic purposes. We then chose eight strains to further understand prophage induction kinetics and host ranges. Following mitomycin C (MMC) induction, we verified nine actively excising prophages through PCR and deep sequencing analysis. Analysis of the induced lysates revealed that four strains contained prophages capable of producing virion particles, and two of these produced phages capable of plaquing on strains in the collection. Our study expands the repertoire of A. baumannii-infecting phages with potential therapeutic applications and provides insights into prophage replication dynamics.
Results
Prophages are abundant in A. baumannii isolates
Our prophage discovery pipeline uses two programs to identify integrative genomic elements (IGEs), which includes prophages. TIGER uses a comparative genomics approach to identify IGEs that contain either tyrosine or serine integrases across the genome,32,34,56 while Islander detects IGEs encoding a tyrosine integrase, which target tRNA or tmRNA genes for integration. 16 We analyzed a curated set of 891 A. baumannii genomes, including 100 strains from a recently isolated panel of diverse strains and the rest from a global clinical study on carbapenem-resistant A. baumannii facilitated by the Antibacterial Resistance Leadership Group13,51 (Supplementary Table S1). TIGER and Islander predicted 789 total prophages among all tested strains, with individual strains containing zero to five prophages (Fig. 1 and Supplementary Table S2). Approximately one-third of strains contained no prophages (323/891, 36.3%), while only 18.1% harbored two or more prophages (161/891). TIGER and Islander also identified integration sites (bacterial attachment site; attB) for putative prophages. Analysis of unique attB sites revealed that more than half of the predicted prophages integrated into tRNAs and other noncoding RNAs (51.3%), while the remaining integrated into protein-coding genes (48.7%, Fig. 2).
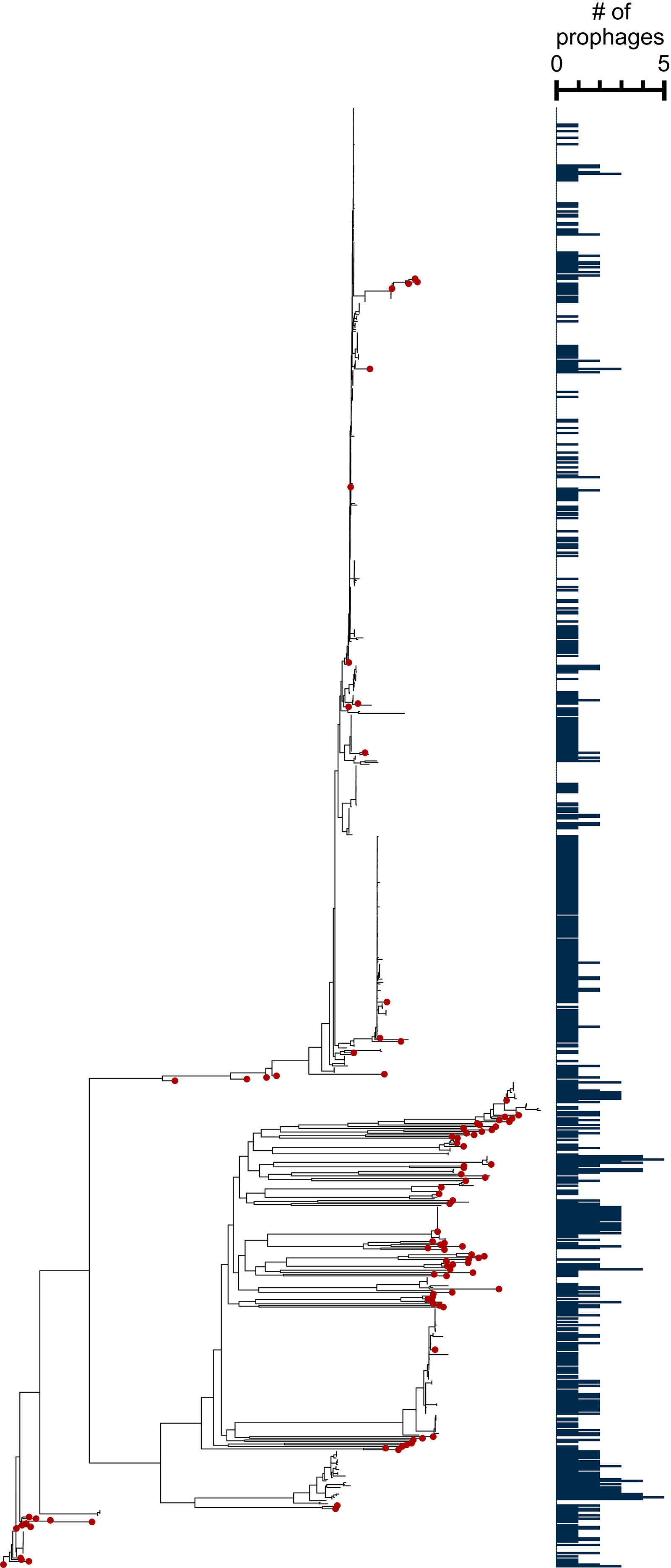
A. baumannii strains form distinct phylogenetic groups, with varying amounts of putative prophages in each genome. Strains are plotted on a mid-rooted core genome tree to represent the diversity of the 861-genome dataset. The bar plot (right) indicates the number of prophages each strain has, from zero to five prophages. Red dots indicate strains from the Galac et al. 13 dataset.
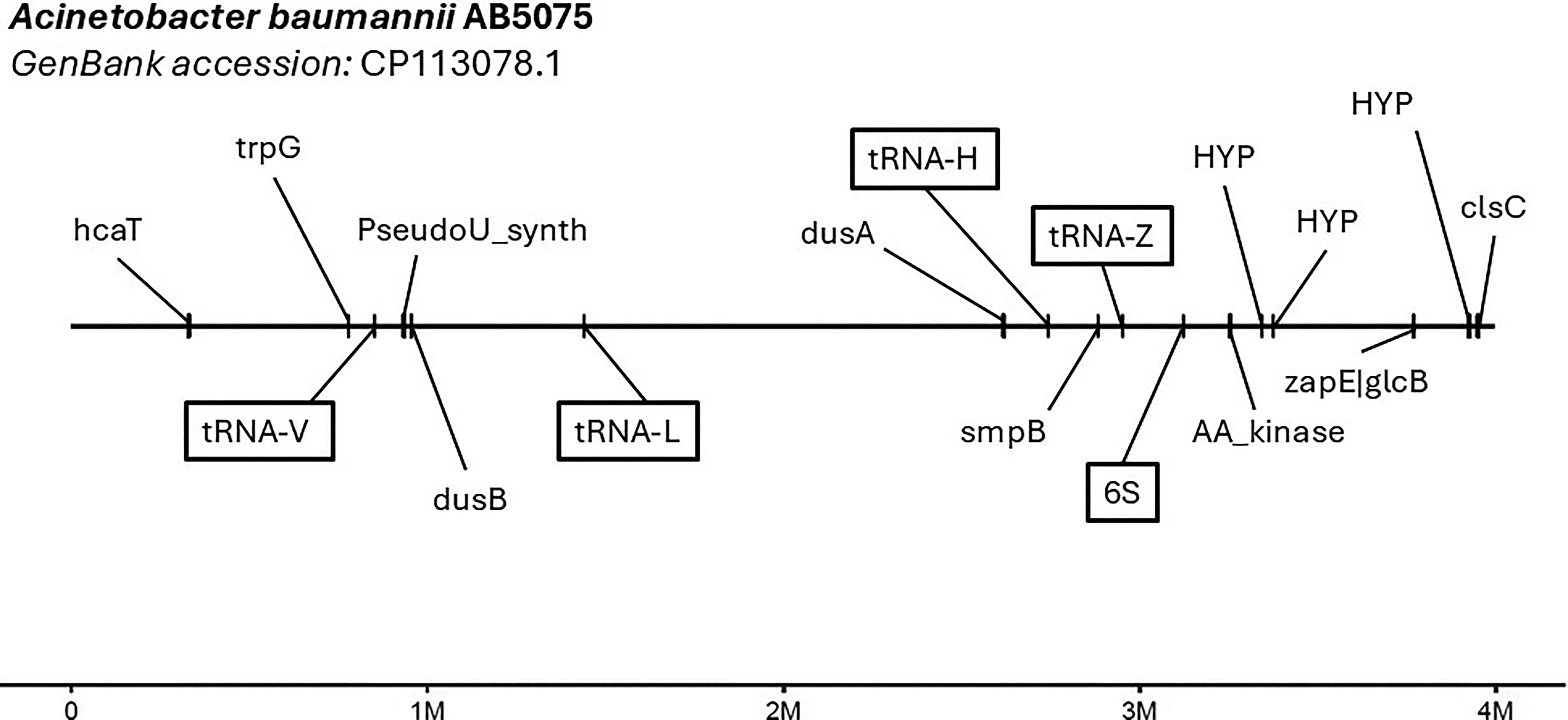
Chromosome map featuring unique integration sites (attB) for predicted prophages, superimposed onto the genome assembly of A. baumannii AB5075 (GenBank accession: CP113078.1). Integration sites used by predicted prophages are labeled with the region that the site corresponds to, with boxed labels indicating sites of nonprotein-coding features such as tRNAs and rRNAs.
To assess the diversity of the predicted prophages and determine whether they expand the current phage repertoire for A. baumannii, we clustered them with 508 A. baumannii phages deposited in National Center for Biotechnology Information (NCBI) GenBank. Clustering analysis revealed 42 total clusters, of which 14 contained prophages. The TIGER/Islander-predicted prophages tend to cluster together, with most exhibiting mosaic genomic architecture (Fig. 3). Some phages that clustered with prophages lacked an annotated integrase. However, additional analysis identified recombinases and/or repressor proteins in these sequences (see Methods; Supplementary Table S3). These predicted prophages substantially increase the known diversity of A. baumannii-infecting phages beyond what has been discovered through traditional isolation methods.
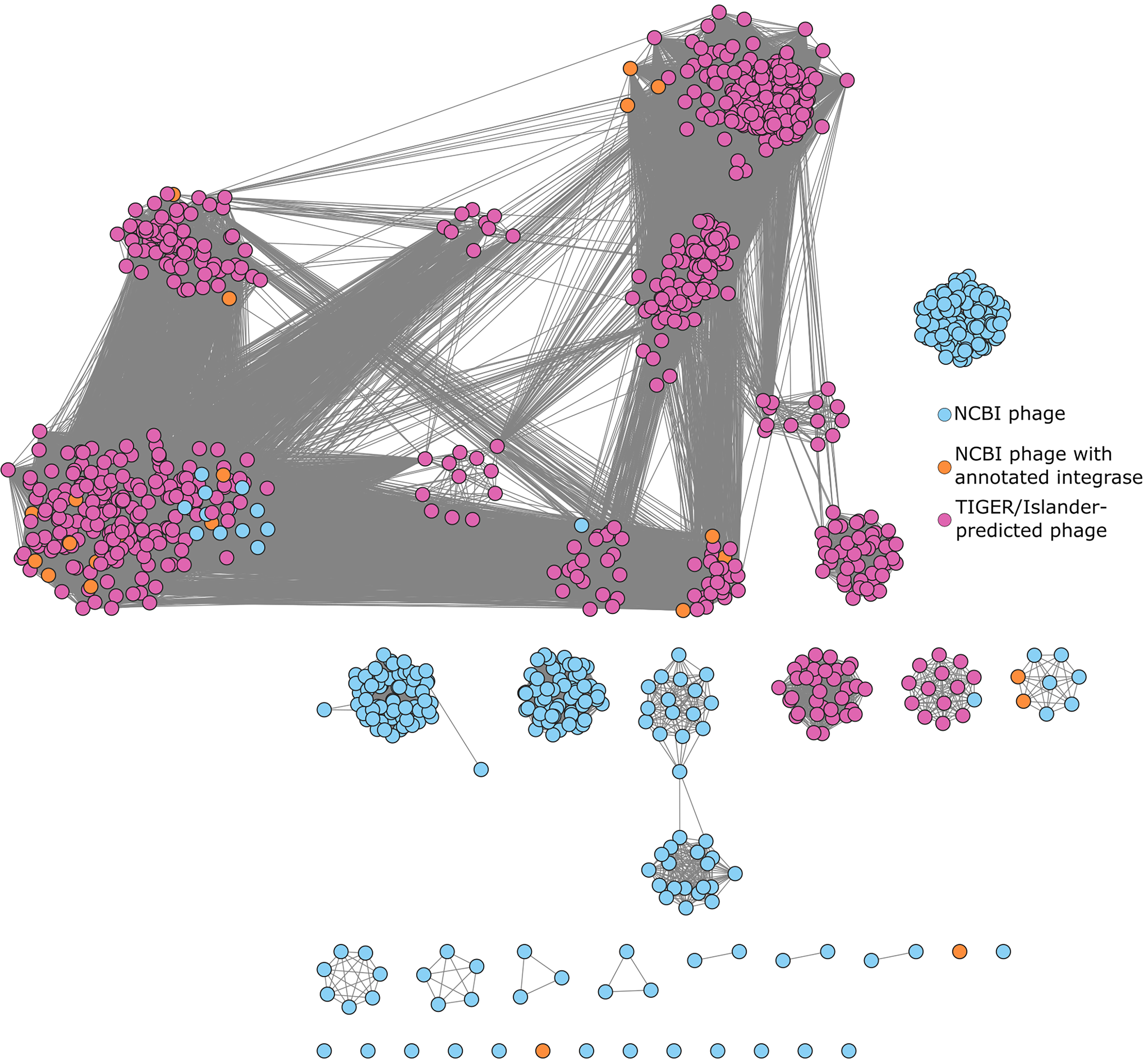
Prophages in A. baumannii form many related clusters. Clusters are computed via the Markov Cluster Algorithm using percent amino acid identity and percent shared genes. 40 TIGER/Islander-predicted prophages have purple labels. NCBI phages that contain any gene already labeled as an “integrase” have orange labels. All other NCBI phages are labeled blue. Clusters were used to determine prophage candidates for further screening by eliminating candidates that were likely to be identical or highly similar.
MMC induction reveals active prophages from nine different strains with variable induction kinetics
We selected 11 strains for further analysis of prophage induction and activity based on the following criteria. The predicted prophages needed to be 40–60 kbp in size, contain a complete set of genes expected for functional phages (including terminase, capsid, tail, portal, and lysin proteins), and represent different clusters.
Typically, a decrease in OD several hours after MMC treatment is seen due to cellular stress, activation of the SOS response, or host cell lysis due to prophage induction.14,25,49 Eight of the 11 strains tested showed a decrease in OD after at least 4 h of MMC treatment (Supplementary Fig. S1). Because TIGER and Islander precisely predict prophage boundaries, we were able to amplify the phage attachment (attP) site, a recombination site formed by circularization of the excised prophage, for nine prophages across the eight strains (Fig. 4). Only one predicted prophage failed to show evidence of excision through attP site detection (2575.42.V).
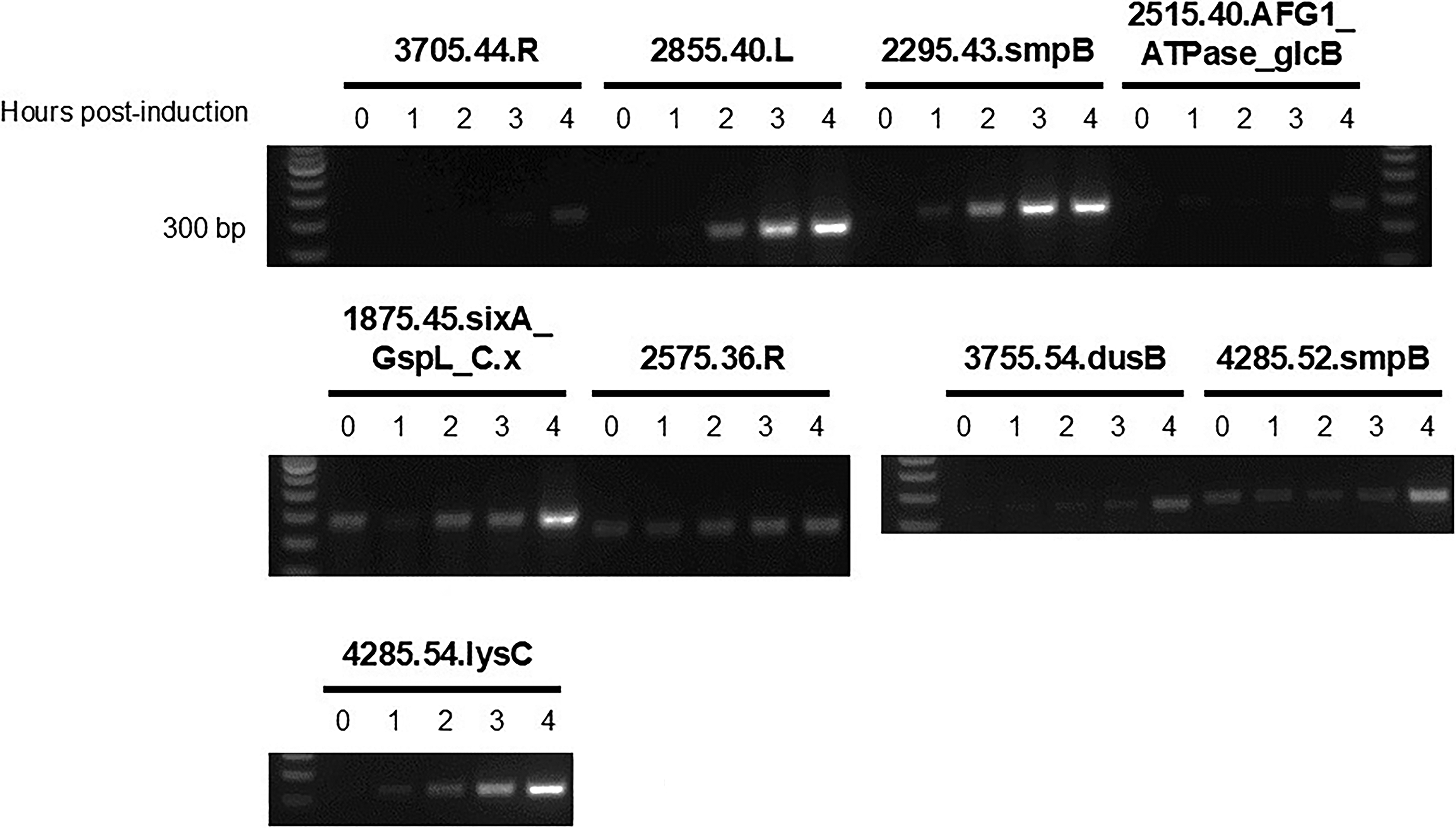
Detection of TIGER-predicted prophage in A. baumannii supernatants. One percent agarose gels showing PCR products (200–300 base pairs) amplifying the attP site from DNase-treated A. baumannii lysates. Prophages are named after the last four digits of their GCA accession number (all accession numbers begin the same; GCA_00649XXXX), the size of the prophage in kilobases, and their integration site. Therefore, 3705.44.R is a 44 kbp prophage found integrated into the arginine tRNA in A. baumannii MRSN30000 (GCA_006493705). A positive attP site band suggests the presence of actively replicating prophage. Note that one strain has more than one prophage detected by PCR (e.g., GCA_006494285).
We spot tested lysates from these strains on 19 A. baumannii strains, including the 11 prophage-laden strains from our initial set plus 8 prophage-free close relatives. MMC-induced lysates from two strains produced plaques (Supplementary Fig. S2). The two strains were found to have only one experimentally validated active prophage each (6492575.36.R and 6492855.40.L), so the plaques were likely to have been caused by the validated prophages. Interestingly, both strains infected by these induced prophages themselves contain at least one putative prophage. Typically, prophage-containing strains prevent secondary phage infections through superinfection exclusion, usually through a prophage-encoded repressor protein.4,11,15
We further characterized the induction biology of these prophages by deep sequencing the MMC-induced genomic DNA to reveal excision and replication kinetics for the nine active phages identified in our PCR screen (Fig. 5). This method allowed us to determine differences in induction timing and replication rates for each prophage, as well as validate additional putative prophages in the same genomes that were originally excluded during selection. Sequencing data confirmed that both predicted prophages in strain GCA_006494285 (52.smpB and 54.lysC) underwent excision, but their induction timing differed. Prophage 52.smpB induced early with peak excision at 3 h post-induction, while 54.lysC induced more slowly and began peaking at 4 h post-induction. Additionally, reads mapping to the attP site of 52.smpB were four times more abundant than reads mapping to its corresponding attB site at 3 h post-induction, which indicates phage replication. Similar trends were observed for strains that only contain one prophage, with phage genome replication verified to occur between 2 and 4 h post-induction (Fig. 5). While the PCR results confirmed that the predicted prophages were actively excising, sequencing data revealed that the intensity and timing of induction varied among different phages.
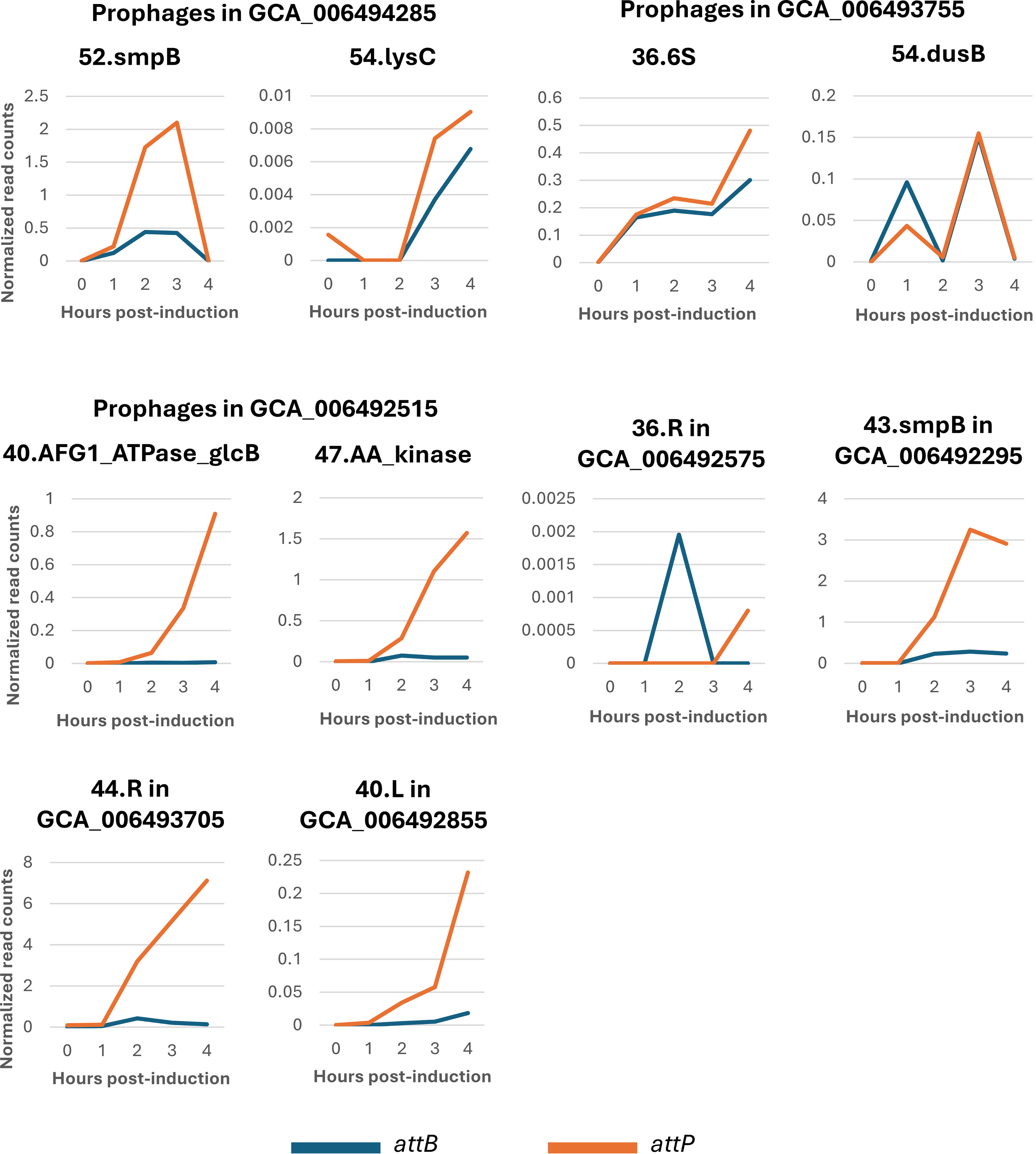
Short-read sequencing data reveals that prophages in A. baumannii undergo different induction dynamics. Line graphs depict the normalized abundances of reads that map to attB (junction left after prophage excision) versus those that map to attP (excised, circularized phage genomes). Read counts are normalized to the quantity of reads that map to attL and attR. Note that y-axes scales are different for each graph, and some genome assemblies contain more than one prophage.
We note that a few of the prophages (54.lysC and 36.R) have extremely low normalized read counts (Fig. 5). One benefit of our method is the precise prediction, which allows us to assess very-low-frequency events, which would otherwise be missed in bulk screening of prophage reads. While these normalized read counts are low, there is still relevant biology to be garnered. We can see the different induction timing between strains with multiple prophages. For example, 52.smpB peaks at 3 h, while 54.lysC begins to induce at 3 h and appears to continue beyond 4 h, a trend that is also seen using PCR (Fig. 4).
Transmission electron microscopy reveals phage particles in induced A. baumannii supernatants
Many of the predicted prophages showed genomic evidence of excision, but only two produced plaques. To verify whether these prophages could produce functional virions, we used transmission electron microscopy (TEM) to visualize phage particles in the MMC-induced lysates. We tested eight MMC-induced lysates and detected intact phage particles in four of them (Fig. 6). All particle-containing lysates contained phages with icosahedral heads. Phages in the supernatants of strains GCA_006493705, GCA_006491875, and GCA_006492855 had consistent capsid diameters ranging from 53 to 70 nm and tail fiber lengths ranging from 162 to 302 nm (Fig. 6A–C and Supplementary Table S7). Notably, lysates from two strains (GCA_006492575 and GCA_006492855) contained phage particles with unique morphologies. Virions from GCA_006492575 possessed tails with unusual protein structures attached (Fig. 6C), while GCA_006492855 contained multiple phages with round capsids attached to long, thin pilus-like structures (Fig. 6D). Unfortunately, these phages could not be propagated from single plaques, making them difficult to isolate.
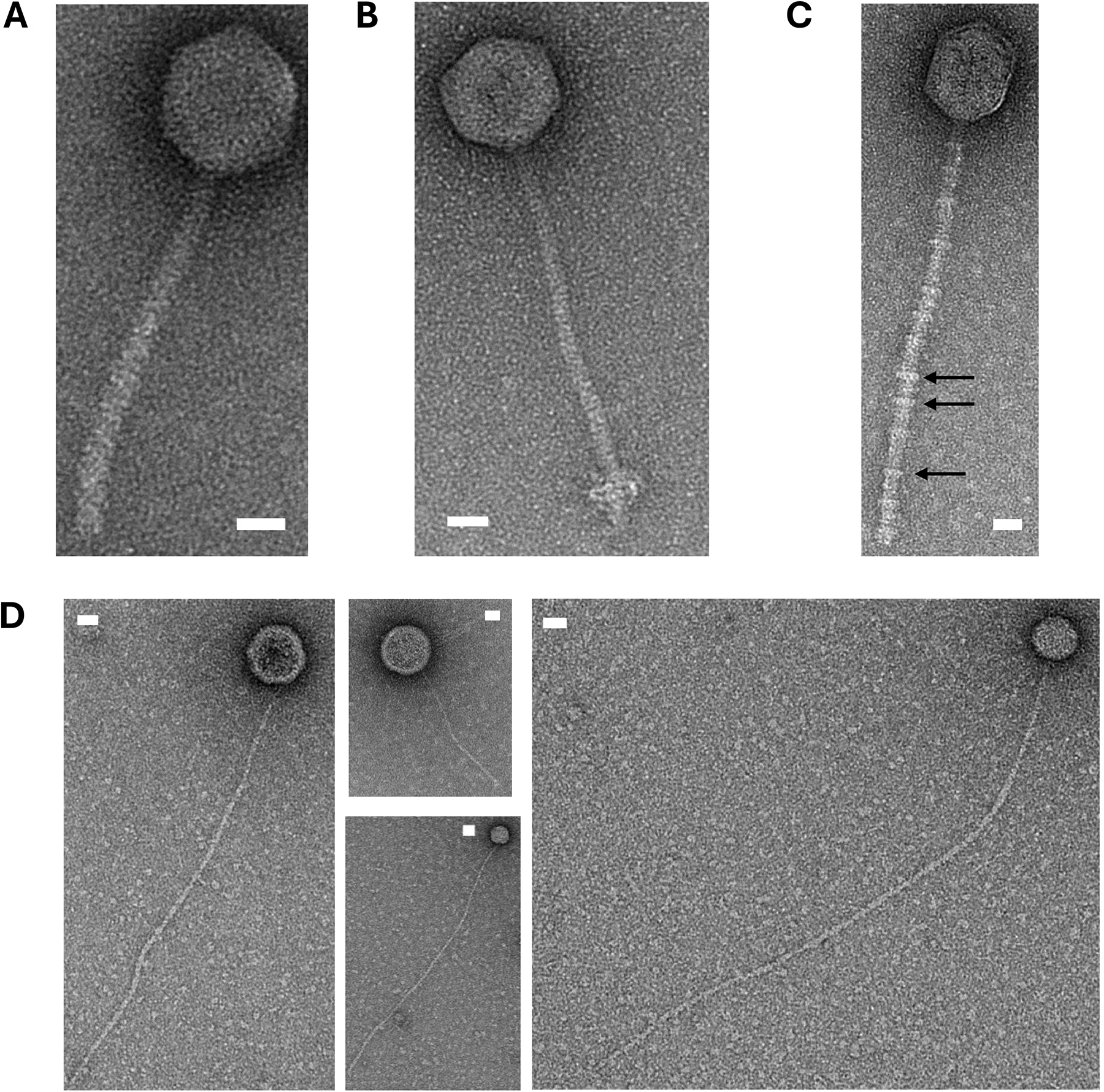
Visualization of phage particles from supernatants from A. baumannii strains using transmission electron microscopy. Phage particles in each panel are from MMC-induced supernatants of the following strains:
Discussion
In this study, we used computational tools to precisely predict prophage within A. baumannii genomes and characterized their activity using culture-based and molecular-based methods. While other studies have examined the diversity of prophages within A. baumannii,9,30,31,47 relatively few temperate phages have been evaluated for therapeutic use.35,41 Furthermore, very few studies have investigated the dynamics of MMC-induced prophage activation.12,33 Here, we precisely predicted multiple prophages from A. baumannii that are unique compared to isolated phages deposited in GenBank. We have further characterized phage excision, replication, and virion production using multiple assays to determine if prophages would be useful as potential therapeutic agents. Our analysis of prophage population dynamics in strains containing one or two prophages reveals the complexity of prophage excision timing and replication kinetics.
While other recent efforts have predicted high numbers of prophages,9,30,31,47 these studies typically used gene content analysis methods that do not precisely characterize the attachment sites for individual prophages. Our pipeline has strict requirements that the predicted prophages contain an integrase gene, a defined attachment site, and a full gene complement of phage genes. These strict requirements reduce the number of prophages predicted but increase the quality of the output. This was observed when comparing our prophage predictions against PHAge Search Tool with Enhanced Sequence Translation predictions for the strains we experimentally tested (Supplementary Table S8). All prophages not predicted by TIGER either lacked an integrase, which may prohibit excision, or did not contain proper attachment sites, which may mean the prophage damaged the host upon entry. TIGER predictions are more likely to be active phages that would be useful in therapeutic applications. Further, our pipeline allows us to easily find prophages that span multiple contigs, which is typically more difficult for popular programs. The precision enables detailed investigation into integration site usage, infection kinetics, polylysogeny dynamics, and prophage activity. We demonstrate that 84.6% (11/13) of predicted prophages from our selected strains were capable of genomic excision. Of these, four produced enough phage particles for TEM visualization, and two produced plaques on strains within our collection. We show prophages are readily inducible and capable of producing virions and infecting strains. In contrast to traditional environmental isolation, many samples typically yield no phage isolates for a specific strain of interest.19,29
The deep sequencing analysis highlights the diversity of induction kinetics across strains and even within the same strain. These results show that prophages can respond to the same stressor at different times, likely coordinating these events to allow both prophages to persist in the population. These types of studies also may allow for future work to understand how the kinetics of prophage induction enhance virulence. These findings are essential for evaluating the therapeutic potential of prophages, either from their native temperate form where bacterial lysis occurs upon stress induction or as engineered virulent derivatives.
To use prophages as therapeutic agents, they must be engineered into virulent phages by removing integrases, repressors, and operator sequences,11,22,24,33 and then further characterized in vivo for safety and efficacy.39,42 Compared to other Gram-negative bacteria, A. baumannii presents two major challenges for effective phage engineering. First, even though clustered regularly interspaced short palindromic repeats (CRISPR)-based phage engineering technologies have been established in many other Gram-negative bacteria,6,17,26 such methods in A. baumannii are still being developed.52,53 Additionally, highly efficient CRISPR enzymes remain limited in this species. Second, while it is possible for phages to be engineered through synthesis and/or transformation of engineered phage genomic DNA (also known as phage “rebooting”),2,20,27 very few A. baumannii phages have been successfully rebooted in model systems like E. coli and have shown reduced efficiency compared to other Gram-negative phages attempted. 7 This highlights the need to understand which phage features enable successfully rebooting. As phage therapy advances, developing methods for engineering and rebooting in nonmodel systems becomes increasingly important. While further research is needed to successfully engineer and deploy prophages as therapeutics, we demonstrate the increased diversity these prophages provide and their potential utility for phage therapy applications.
Materials and Methods
Prophage prediction, clustering, and host strain phylogeny in A. baumannii
Prophage prediction was performed as described previously. 33 Briefly, two discovery platforms for IGE, Islander 16 and TIGER, 32 were applied to 891 A. baumannii genomes from GenBank. Genomic islands were annotated with our Tater software. 32 The software uses Prodigal 18 to call open reading frames; Prokka and Pfam-A Hidden Markov Model databases were used to predict phage protein functions. IGEs are sorted into different categories based on the type of island. Briefly, 789 phages were identified as “Phage 1,” which are prophages with a credibly complete genome, while 289 phages were identified as “Phage 2” because they may be missing some phage genes. The software detected only five putative filamentous phage. All candidates identified as “Phage 1” are further filtered based on genome size and whether these phages are present in unique clusters.
A core genome tree for all A. baumannii strains was built with IQ-TREE (v.3.0.1) 37 using the ModelFinder algorithm 23 to determine the appropriate substitution model and 1000 ultrafast bootstrap replicates to generate a consensus tree. Prophage counts were mapped onto the resulting phylogenetic tree using the R package ggtree (v.3.10.0). 55 Prophage DNA sequences were then clustered by the Markov Cluster Algorithm based on an average amino acid identity (>40%) and minimum percent of genes shared (>20%; 40 https://github.com/snayfach/MGV/blob/master/aai_cluster/README.md) to select phages for screening by minimizing the number of phages in the same cluster screened. Phages from NCBI were quickly screened for integrases with a keyword search, and other phages from NCBI that clustered with TIGER/Islander-predicted prophages were evaluated further with HMMER and HMMs from the PHROGs dataset (https://www.ebi.ac.uk/Tools/hmmer/search/phmmer) 48 for integrases and repressor proteins.
Prophage induction and plaque assays
For the initial screening of prophage-laden strains, A. baumannii strains were grown overnight at 37°C in Luria broth (LB) broth while shaking at 225 Revolutions per minute. Overnight cultures were back-diluted 1:100 into 150 uL LB in sterile 96-well plate wells (Fisher Scientific #08-772-54) before placing the plate in a Tecan Spark® Cyto plate reader at 37°C with an orbital shaking intensity of 3. Optical density was measured every 10 min. When cultures reached an OD600 of 0.4–0.5, cells were induced with 1 μg/mL MMC (Fisher Scientific# BP25312) and incubated for up to 6 h (Supplementary Fig. S1). Three technical replicates were used for each strain, and graphs were generated in Microsoft Excel showing the average of each well per timepoint measured. Eight prophage-containing strains with selected prophages were selected for further analysis.
To collect lysates and genomic DNA, A. baumannii strains were grown overnight at 37°C in LB broth, back-diluted 1:100 in 5 mL LB, and incubated to an OD600 of 0.4–0.5. Cultures were then induced with 1 μg/mL MMC (Fisher #BP25312). Then, 1 mL aliquots were collected from cultures at 0, 1, 2, 3, and 4 h post-induction. Cells from aliquots were pelleted by centrifugation at 6000× g for 10 min. Supernatants were harvested and filtered through a 0.2-μm syringe filter (Whatman® #WHA9916-1302) before DNaseI treatment. Cell pellets were stored at –20°C for subsequent genomic DNA extractions. Plaques were generated either by mixing filtered supernatants with 4 mL of 0.6% (soft) agar and 20 μL of host bacterial culture or by spotting 3 μL supernatant onto host lawns made with 4 mL soft agar and 20 μL of host bacterial culture. Plates were incubated overnight at 37°C to visualize plaques on host lawns.
Assessing prophage activity with PCR and deep sequencing
One output of the TIGER program is a set of predicted att sites indicating the recombination sites of the integrated or replicating prophage (attL, attR, attB, and attP). 32 Primers to test for actively replicating phage were designed according to the att sites computed for each prophage (Supplementary Table S5). Supernatants from prophage induction treatments were then DNaseI-treated (Norgen Biotek #25710) to remove DNA not protected by capsids. DNaseI treatments consisted of adding 10 μL DNaseI per 1 mL of supernatant and incubating the supernatants for 15 min at room temperature before deactivating the DNaseI at 75°C for 5 min. Then, 2 μL was used as a template for PCR detection of attB and attP sites, indicative of excised and circularized prophage genomes. PCRs were set up using the Quick-Load® Taq 2X Master Mix (New England BioLabs # M0271L) following manufacturer directions for PCR cycling times and annealing temperatures. Control PCRs were run to confirm primers only detected expected templates (Supplementary Fig. S3).
To explore prophage dynamics, we used deep sequencing. Genomic DNA was isolated from cell pellets using the Qiagen DNeasy blood and tissue kit (Qiagen #69504). Sequencing libraries from genomic DNA were prepared using the Illumina DNA Prep (M) Tagmentation kit (Illumina #20060059) and using Illumina® DNA/RNA UD Indexes Set B (Illumina #20091656) following the manufacturer’s recommended protocol. DNA libraries were quantified using the Qubit High-Sensitivity dsDNA Quantitation kit (Thermo Fisher #Q32851) and pooled in equal quantity to make a final library. Quality control on the final library was performed with the Qubit quantitation kit and 4150 TapeStation System (Agilent #G2992AA) using the High Sensitivity D5000 Reagents and ScreenTape (Agilent #5067-5592 and #5067-5593). Samples were either shipped to Genewiz for Illumina sequencing or sequenced in-house on a NextSeq™ 2000 (Illumina) system using the NextSeq™ 2000 P3 XLEAP-SBS™ Reagent Kit (200 Cycles; cat # 20100989), generating an average of 15 million reads per sample to ensure quantification of low-abundance reads. Reads were processed through Juxtaposer, 44 a software package built to detect genomic recombination in sequencing reads. We generated probes for attB, attP, attL, and attR to detect and count recombinant reads (Supplementary Table S6).
TEM imaging of phage
Supernatants from induced bacterial cultures were concentrated by spinning samples down at 2300× g for 4 h at 4°C before shipping 50 μL of the supernatant to the University of Maryland, Baltimore County, for further analysis. Imaging work was performed by Tagide deCarvalho at the Keith R. Porter Imaging Facility at UMBC on a Hitachi HT7800. Capsid diameters and tail lengths were measured with ImageJ software using a provided scale bar as a reference.
Authors’ Contributions
Conception and design of the work were contributed by J.T., V.K.M., and C.M.M. J.T. executed experiments and performed most of the bioinformatic analyses. C.M.M. supervised the work and supported bioinformatic analyses. A. baumannii strains and genome assemblies were curated and provided by V.K.M., J.T., and C.M.M. wrote the article. All authors read, commented on, and approved the final article.
Footnotes
Acknowledgments
The authors thank Alexey E. Kazakov (Lawrence Berkeley National Laboratory) for assisting with the curation of A. baumannii genomes for this project. They also thank Kelly P. Williams and Ellis L. Torrance and Hannah McClain (Sandia National Laboratories) for their work on the Phage Factory pipeline, Juxtaposer, and att site software (article in prep). They thank Joanna Steczynska (Sandia National Laboratories) for her thoughtful feedback on this article.
Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC (NTESS), a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration (DOE/NNSA) under contract DE-NA0003525. This written work is authored by an employee of NTESS. The employee, not NTESS, owns the right, title, and interest in and to the written work and is responsible for its contents. Any subjective views or opinions that might be expressed in the written work do not necessarily represent the views of the U.S. Government. The publisher acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this written work or allow others to do so, for U.S. Government purposes. The DOE will provide public access to results of federally sponsored research in accordance with the DOE Public Access Plan.
Data Availability
Genome assemblies from A. baumannii strains used for the initial bioinformatics analysis are described in Supplementary Table S1. Acinetobacter-infecting phage accessions from NCBI are described in Supplementary Table S4.
Author Disclosure Statement
The authors declare no potential conflicts of interest.
Funding Information
This was completed as part of the BRaVE Phage Foundry, which is supported by the U.S. Department of Energy, Office of Science, Office of Biological & Environmental Research under contract number
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.