
Abstract
Artificial intelligence (AI) is transforming every stage of oncology drug development, offering unprecedented opportunities for innovation, efficiency, and patient-centered care. In January 2026, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) jointly published the “Guiding Principles of Good AI Practice in Drug Development” (“FDA-EMA Guiding Principles”). 1 These 10 principles establish a shared transatlantic framework for the responsible design, validation, and oversight of AI systems across the drug lifecycle. This article distills the regulatory intent behind the FDA–EMA Guiding Principles and translates them into operational guidance for oncology researchers, clinicians, and institutional leaders engaged in evidence generation.
Introduction: Artificial Intelligence as an Evidence-Generating System in Oncology
Artificial intelligence (AI) now influences biomarker discovery, patient stratification, trial design, imaging interpretation, dose optimization, manufacturing controls, and postmarket safety analytics. In oncology, where endpoints are often complex and patient populations heterogeneous, AI is increasingly embedded in the evidentiary backbone of development programs.
Until recently, regulatory expectations for AI systems were diffuse. The Food and Drug Administration–European Medicines Agency (FDA–EMA) Guiding Principles mark a pivotal moment: They signal regulatory convergence across two leading authorities and establish clear expectations for governance, validation, lifecycle oversight, and transparency. Although framed as high-level principles, they function operationally as a regulatory readiness checklist.
For oncology investigators and clinicians, the message is clear. When AI systems influence evidence that may support regulatory approval—particularly primary or key secondary endpoints—regulators will expect documentation, validation, and quality controls comparable to other critical development systems. Deficiencies in AI governance or validation may result in information requests, review delays, or heightened scrutiny during marketing application assessment. AI oversight can no longer be siloed within data science teams; it must be embedded within institutional research governance and quality systems.
The 10 principles can be organized into 4 strategic categories that translate regulatory expectations into operational practice for oncology drug development.
Category 1: Governance and organizational foundations
Principles 1, 2, and 5—Human-Centric Design, Risk-Based Approach, and Multidisciplinary Expertise—establish the organizational infrastructure for responsible AI use.
AI must augment, not replace, scientific and clinical judgment. Risk stratification should determine oversight intensity based on how AI outputs influence regulatory submissions. Applications generating primary endpoints, stratifying pivotal trial populations, or supporting safety determinations warrant heightened governance and validation. Lower-risk tools, such as literature screening or early hypothesis generation, require proportionate controls.
Key operational actions in oncology settings include the following:
Establishing AI governance committees with representation from discovery research, translational science, clinical development, biostatistics, regulatory affairs, quality assurance, pharmacovigilance, data science, and legal/ethics leadership. Classifying AI applications by regulatory impact and defining oversight pathways accordingly. Embedding AI review into stage-gate development processes. Defining clear human oversight checkpoints before AI outputs are incorporated into regulatory submissions.
For academic medical centers and comprehensive cancer centers conducting industry-sponsored trials, governance also has inspection implications. AI systems influencing protocol design, endpoint generation, or safety monitoring may be examined during GCP inspections. Institutions should, therefore, ensure that AI oversight structures are documented, auditable, and aligned with existing compliance frameworks.
Category 2: Data quality and technical standards
Principles 3, 6, and 7—Adherence to Standards, Data Governance and Documentation, and Model Design and Development Practices—address the technical foundation of regulatory-grade AI.
Regulators expect AI systems to meet established GxP standards where applicable. In oncology, this may include the following:
Training data must meet standards commensurate with their regulatory impact. Data provenance, inclusion/exclusion criteria, consent status, data transfer agreements, and preprocessing steps should be documented in a manner analogous to investigational product controls.
Oncology-specific risks warrant particular attention. AI models trained on limited demographic or tumor subtype datasets may perform inconsistently in broader or underrepresented populations. Imaging algorithms developed using single-center radiology protocols may not generalize across global multicenter trials. Biomarker stratification tools derived from narrow genomic repositories may introduce systematic bias. Regulators increasingly expect systematic bias assessment and documentation of performance variability across clinically relevant subgroups.
Comprehensive technical documentation should include model architecture, training parameters, validation datasets, performance metrics, known limitations, and version control history. These materials should be maintained in formats suitable for regulatory submission appendices.
Category 3: Validation and performance management
Principles 4 and 8—Clear Context of Use and Risk-Based Performance Assessment—ensure that AI tools perform reliably within their defined roles.
Every AI system must have a precisely defined context for its use specifying its function, inputs, outputs, intended population, and decision boundaries. In oncology development, it may include the following:
AI generating or supporting RECIST-based response measurements. Predictive models identifying likely responders to targeted therapies. Algorithms prioritizing adverse event signals.
Validation strategies must align with regulatory impact. AI contributing to pivotal endpoints may require prospective validation under conditions approximating intended use. Analytical validation comparing AI outputs to established reference standards may suffice for exploratory or supportive applications.
Human–AI interaction also requires validation. Institutions should assess whether oncologists, radiologists, pathologists, or trial monitors can appropriately interpret and apply AI outputs. Workflow validation is essential to ensure that the AI system performs not only technically but also operationally.
Validation documentation should be regulatory-ready, including protocols, statistical analysis plans, performance results, subgroup analyses, and limitation discussions. Inadequate validation may lead to regulatory queries during marketing application review or delay the acceptance of AI-derived evidence.
AI-Derived Endpoints: Automated RECIST and Emerging Response Metrics
A particularly high-impact application of AI in oncology drug development is the generation or support of efficacy endpoints derived from imaging—most notably RECIST-based tumor response assessments. Under RECIST 1.1, 5 radiologists manually identify target lesions, measure their longest diameters, and categorize response as complete response, partial response, stable disease, or progressive disease. These determinations directly inform pivotal endpoints such as objective response rate and progression-free survival.
AI systems are increasingly capable of automating lesion detection, segmentation, and measurement, and, in some cases, classifying response categories algorithmically. When AI contributes to or replaces manual measurements underpinning primary or key secondary endpoints, it becomes a high regulatory-impact tool.
In this context, the FDA–EMA Guiding Principles translate into concrete expectations. The context of use must be precisely defined in the protocol and statistical analysis plan, including whether AI operates autonomously or with human confirmation. Analytical validation should demonstrate concordance with expert radiologist assessments across representative tumor types, imaging platforms, and geographic sites. Subgroup analyses are essential to assess performance variability across patient populations and imaging modalities. Algorithm version control is critical; models used to generate pivotal endpoint data should be locked prior to database closure, with documented change control procedures.
For emerging AI-derived metrics—such as volumetric tumor burden or composite radiomic response indices that extend beyond traditional RECIST criteria—substantial analytical validation and clinical outcome correlation may be required to demonstrate reliability, reproducibility, and regulatory comparability to established endpoints. If AI-derived volumetric or radiomic endpoints are intended to replace or function equivalently to established response criteria, such as RECIST, sponsors may need to engage formal biomarker qualification pathways to support regulatory acceptance.
In certain configurations, AI systems used to derive oncology endpoints may also implicate medical device regulatory frameworks. Where AI functions solely as an internal research tool to generate or support trial endpoints—without influencing real-time patient management—it may not require independent market authorization or evaluation as software for a medical device. However, if the AI is intended to guide treatment decisions, determine progression triggering therapy changes, or be commercially distributed for diagnostic use, medical device regulation under U.S. or EU law may apply. Even where formal device authorization or certification is not required, regulators are likely to expect device-like controls, including robust validation, version locking, change management, and quality system oversight. As AI-derived endpoints become central to efficacy determinations, sponsors should anticipate increasing convergence between drug and software regulatory expectations.
Category 4: Lifecycle monitoring, transparency, and regulatory integration
Principles 9 and 10—Lifecycle Management and Clear, Essential Information—recognize that AI systems are dynamic and require continuous oversight.
In multiyear oncology programs, patient populations evolve, standards of care shift, and diagnostic technologies advance. AI models may require revalidation in the following cases:
Standard-of-care therapies alter baseline patient characteristics. Diagnostic criteria or response assessment guidelines are updated. Geographic expansion introduces new population variability. Model updates or retraining occur.
Lifecycle management should integrate AI into existing pharmaceutical quality systems, including change control, deviation management, CAPA processes, and internal audits.
For programs extending into postapproval phases, institutions should anticipate how AI model updates interact with supplemental applications, label expansions, or postmarketing commitments. Transparent documentation of model modifications and revalidation analyses will be essential if AI-derived evidence continues to support regulatory claims.
Transparency also extends to regulatory engagement. Sponsors and investigators should disclose AI use during regulatory meetings, provide clear summaries of functionality and limitations, and include structured technical documentation within submissions. The objective is to enable regulatory reviewers to assess what the AI does, whether validation is adequate for the claimed context of use, and how it may influence regulatory decision-making.
Practical Considerations for Principal Investigators and Translational Researchers
For clinicians and academic investigators, several practical questions may serve as an internal governance checklist as follows:
Is the AI model’s context of use clearly described in the protocol? Has the model been externally validated in a population comparable to the trial cohort? Are model updates frozen during pivotal trial phases? Is there a documented plan for monitoring performance drift? Are data provenance and subgroup performance analyses documented in a format suitable for regulatory review?
Embedding these considerations early in protocol development can reduce downstream regulatory risk and strengthen evidentiary credibility.
From Principles to Practice: Building Institutional Readiness
Operationalizing the FDA–EMA Guiding Principles requires institutional commitment (Fig. 1) as follows:
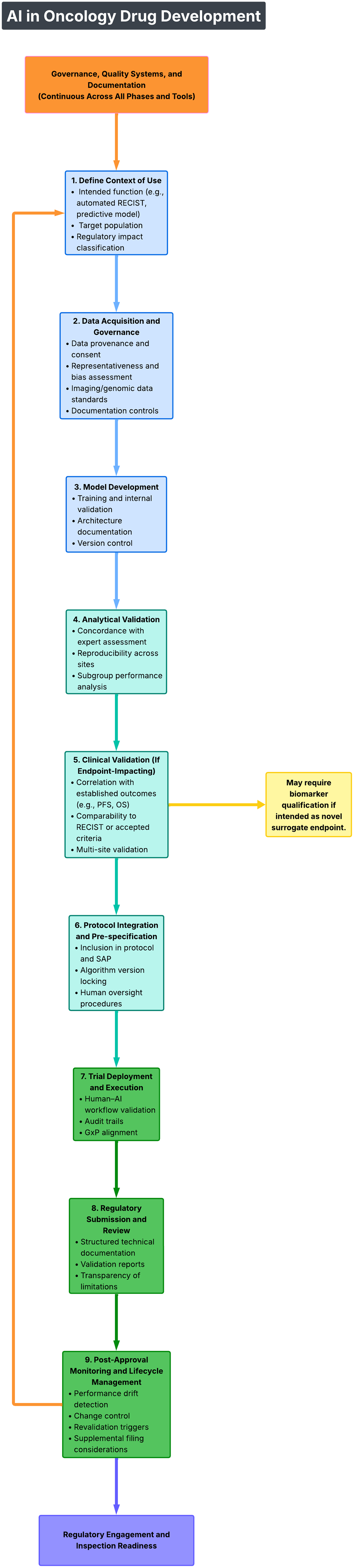
Artificial intelligence in oncology drug development.
Build and maintain a comprehensive AI inventory documenting context of use, risk classification, validation evidence, and accountable leadership.
Establish cross-functional governance infrastructure with defined decision-making authority.
Integrate AI oversight into GxP-compliant quality management systems.
Invest in bidirectional AI literacy—training clinicians on AI fundamentals and data scientists on regulatory and oncology development standards.
Engage regulators early when deploying novel or high-impact AI systems.
These principles also align with broader global AI governance trends. Institutions operating internationally should anticipate increasing convergence between drug regulatory expectations and cross-sector AI governance frameworks.
Conclusion: From Compliance to Regulatory and Scientific Excellence
The FDA–EMA Guiding Principles mark a new phase in AI-enabled oncology drug development—one defined by harmonized expectations, governance maturity, and operational accountability. They are not aspirational ideals; they represent the emerging baseline for regulatory-grade AI.
Organizations that treat these principles as a structured governance roadmap will be positioned not only to mitigate regulatory risk but also to enhance evidentiary rigor and patient trust. In oncology, where therapeutic decisions carry profound consequences, robust AI governance is not merely a compliance obligation—it is a scientific and ethical imperative.