
Abstract
Aortic valve stenosis (AS) is the most prevalent form of valvular heart disease in developed countries. However, a limited understanding of its molecular pathogenesis has hindered the development of an approved pharmacological therapy. Currently, the only effective treatment is aortic valve replacement once the disease becomes severe. This review synthesizes our current understanding of AS with a multiomics approach, which integrates findings from genomics, transcriptomics, proteomics, and metabolomics to construct a systems-level view of AS. Recent landmark studies in 2025 have exponentially expanded the known genetic architecture of AS, identifying over 240 risk loci through massive multiancestry meta-analyses and deep learning–derived imaging phenotypes. Genomics has identified key genetic risk loci, most notably LPA, and established that the genetic framework of AS is distinct from that of atherosclerosis. Transcriptomics, particularly at the single-cell level, has revealed cellular heterogeneity within the valve, identifying specific profibrocalcific cell populations and highlighting pathogenic processes such as endothelial-to-mesenchymal transition. This discipline has also revealed the role of noncoding RNAs, such as H19, in regulating osteogenic programs. Proteomics has identified effector proteins and candidate circulating biomarkers, including Matrix Metalloproteinase-12 as a marker of progression and Complement C1q TNF-Related Protein 1 as a potential causal factor. Additionally, proteomics has uncovered a novel link between AS and amyloidogenesis. Metabolomics has mapped the metabolic reprogramming that fuels AS, identifying procalcific metabolites such as lysophosphatidic acid and linking them to genetic risk factors. These omics domains reveal cohesive pathogenic pathways, such as the Lp(a)-autotaxin-LysoPA axis. This comprehensive molecular characterization has revolutionized our understanding of AS, reframing it as an active, genetically influenced disease and paving the way for the development of targeted pharmacotherapies and precision diagnostics.
This is a visual representation of the abstract.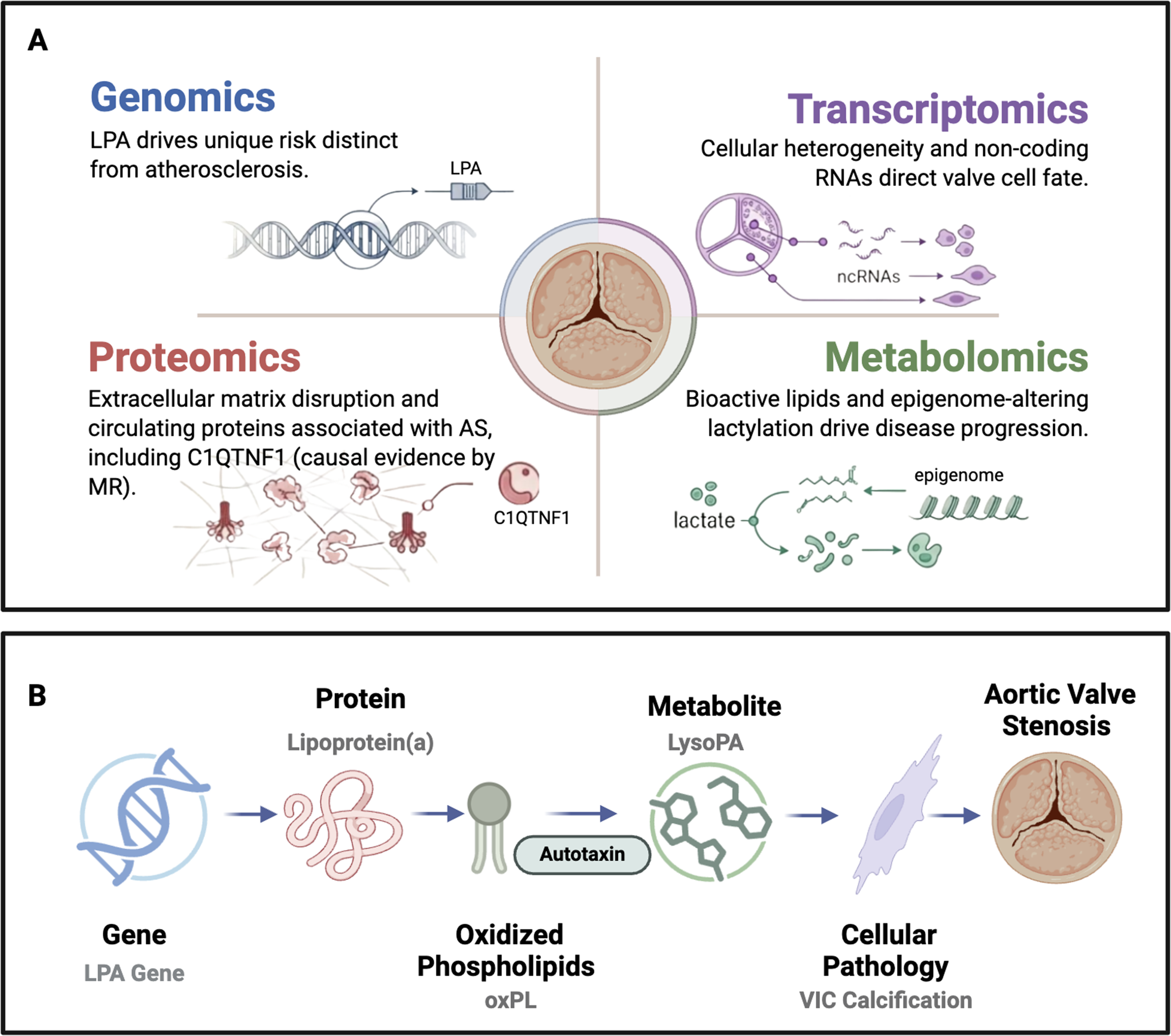
Keywords
Introduction
Calcific aortic stenosis (AS), the final stage of calcific aortic valve disease (CAVD), is a growing public health crisis. It is the most common form of valvular heart disease in developed nations, affecting more than 2% of individuals over the age of 65.1,2 Its prevalence is expected to nearly triple in the coming decades, driven by the aging of the global population and the rise in related conditions such as obesity, type 2 diabetes, and chronic kidney disease.1,3 Once symptoms manifest, the prognosis for severe AS is poor; without intervention, the mortality rate approaches 50% within 2 years.4,5 Despite this clinical urgency, there are currently no approved pharmacotherapies capable of halting or reversing the progression of aortic valve degeneration. Aortic valve replacement (AVR), either surgical or transcatheter, is currently the only effective treatment, which is both invasive and costly. 1
The historical view of AS as a passive, degenerative disease has been replaced by the understanding that AS is an active, complex, and cell-mediated biological process. 6 The contemporary model presents AS as multifaceted; it involves lipid infiltration, chronic inflammation, extracellular matrix (ECM) remodeling, and progressive fibrosis. Importantly, AS involves the osteogenic differentiation of resident valvular cells, which leads to ectopic valve calcification. 5 These interconnected modalities explain the failure of therapies that target single pathways. For example, despite histological similarities between AS and atherosclerotic lesions, large-scale clinical trials of statins, which are highly effective in treating atherosclerosis, have failed to show any benefit in slowing the progression of AS. 7 This finding reflects a fundamental difference in the molecular foundation of the 2 diseases.
A systems biology approach that integrates multiple layers of molecular data can be employed to dissect the complexities of this disease and identify potential therapeutic targets.1,5 Multiomics combines genomics, transcriptomics, proteomics, and metabolomics to provide a holistic framework to explore AS pathogenesis. This review will synthesize relevant findings from each of these omics modalities and explore how their integration can reshape our understanding of AS and guide the development of pharmacological therapies.
Genomics
Genomic studies have identified inherited variants that increase susceptibility and have offered insights into the underlying causal mechanisms of AS.
Landmark Genome-Wide Association Studies Findings
Genome-wide association studies (GWAS) have been pivotal in assessing the genetic framework of the disease. A strong association has been found between AS and the LPA gene on chromosome 6, which encodes apolipoprotein(a). Apolipoprotein(a) binds to apolipoprotein B to form lipoprotein(a) (Lp(a)). 8 In particular, the intronic single-nucleotide polymorphism rs10455872 shows a significant association with both the presence of aortic valve calcification and the clinical diagnosis of AS. 9 Mendelian randomization studies have provided compelling evidence that lifelong, genetically elevated levels of Lp(a) are not merely correlated with but have a causal role in AS. 8 This finding also supports the role of lipid-mediated inflammation as a central pathogenic pathway in AS.
In 2024, a large-scale GWAS conducted by Thériault et al identified 32 genetic regions associated with AS, 20 of which were novel. 10 The study involved a meta-analysis of 14 819 cases and 927 044 controls of European ancestry. Its findings link AS to genes responsible for lipid metabolism (LPL, LDLR), vascular remodeling (PDGFRA, PDE3A), and valve development (PRRX1, TWIST1). Mendelian randomization analyses confirmed tissue-specific causal effects for several identified loci. A notable example is the TWIST1 gene, whose risk-contributing activity was found to be exclusively active in the valve despite the gene's broad expression pattern. 10
The foundational GWAS meta-analyses conducted between 2023 and 2024 by Small et al, Chen et al, and Thériault et al have collectively broadened our understanding of AS pathogenesis, implicating a diverse number of biological pathways. Key genetic loci implicated in AS are summarized in Table 1.
Inflammation and Calcification Pathways
The identification of a risk locus near IL6, the gene encoding the pro-inflammatory cytokine interleukin-6, provides genetic evidence for a causal role of inflammation in AS.8,11
Genetic Regulation and Structural Development
Loci near PALMD (Palmdelphin) and TEX41 (Testis Expressed 41) are strongly associated with both AS and bicuspid aortic valve (BAV), a common congenital anomaly.10,12–14 A shared genetic foundation suggests that AS may not solely be a degenerative condition of aging. Instead, it could be a late-life manifestation stemming from a subtle developmental predisposition. The biological pathways influenced by these genes appear to play a role in both the initial formation of the valve and its long-term structural integrity, indicating that some individuals may be predisposed to degeneration from birth.
Lipid and Fatty Acid Metabolism
Findings have demonstrated that lipid metabolism's role in AS extends beyond just Lp(a). A risk locus identified in the FADS1/2 gene cluster, which is involved in the desaturation of polyunsaturated fatty acids, and another at the CELSR2 locus points to a broader dysregulation of other lipid pathways.8,15 At the CELSR2 locus, the likely causal gene is SORT1. This gene encodes sortilin, a protein that modulates the secretion of very-low-density lipoprotein cholesterol, which further implicates lipid handling in AS pathogenesis. 8
Novel and Pleiotropic Loci
Recent GWAS have uncovered several novel loci with pleiotropic effects, including regions near SLMAP, MECOM, CDAN1, and, most notably, FTO. 8 The FTO locus is widely known for having the largest effect size on obesity. Given that obesity is a clinical risk factor for AS, the simplest hypothesis would be that FTO increases AS risk by increasing body mass index (BMI). 8 However, further analyses have revealed a more complex relationship. The association between the FTO locus and AS remains significant even after statistical adjustment for BMI. 8 Mediation analyses confirm that a substantial portion of the FTO locus’ effect on AS is independent of its effect on adiposity. 8 Additional investigation suggests an alternative mechanism: the risk variant at this locus may influence AS risk by affecting the expression of RBL2. 8 This gene is implicated in telomere regulation and cellular senescence, which suggests that a single genetic locus can influence 2 related diseases (obesity and AS) through distinct mechanistic pathways (adiposity vs cellular aging). 8
Recent landmark studies in 2025 have built upon this prior work. Kany et al applied deep learning to cardiac magnetic resonance (CMR) images from 59 571 UK Biobank participants, deriving 3 quantitative measures of valve function: peak velocity, mean gradient, and aortic valve area. 16 They conducted GWAS on these continuous imaging-derived traits and then combined them with disease-based GWAS using multitrait analysis. This approach identified 166 distinct loci, of which 134 were associated with AS risk. By capturing normal variation in valve function in the general population, the method detects genetic influences on the valve long before clinical symptoms appear. 16 Simultaneously, a massive multiancestry meta-analysis by Small et al involving over 86 000 cases identified 241 independent risk loci. 17 Crucially, this recent work has moved from nomination to functional proof. While CMKLR1 and LTBP4 were not statistically significant in the initial transcriptome-wide association study, Small et al selected them for functional validation based on their biological relevance. Silencing these genes in human valvular interstitial cells significantly decreased mineralization, directly implicating polyunsaturated fatty acid signaling and TGF-β pathways as drivers of AS. 17
Aortic Stenosis Versus Atherosclerosis
One of the most critical insights to emerge from AS genomics is the distinction between the genetic framework of AS and that of atherosclerotic cardiovascular disease (ASCVD). An analysis of 14 replicated lead variants for AS found that only 2 (LPA and CELSR2) were also significantly associated with ASCVD. 8 This supports a consistent clinical observation: therapies developed for atherosclerosis, such as statins, have failed to benefit patients with AS.18,19 Trenkwalder et al found that there is only a moderate genetic correlation (0.15) between the 2 diseases. 20 Out of 17 genetic risk loci for AS, 11 were AS-specific and showed no association with CAD. 20 Genetic evidence now guides research away from simply repurposing atherosclerosis drugs and towards developing unique therapies for AS.
However, the distinctness of AS from atherosclerotic disease should not be overstated. While the bulk of AS-specific genetic architecture does not overlap with ASCVD lead variants, integrative analyses have consistently identified cross-phenotype connections to broader cardiometabolic risk factors, including circulating lipoproteins, blood pressure, and inflammation.10,16 In particular, Mendelian randomization findings on LDL cholesterol have been inconsistent: Small et al found that the apparent causal effect of LDL-C on AS was attenuated when adjusting for Lp(a), suggesting much of the LDL signal may be confounded by Lp(a) carriage, 8 whereas Kany et al found evidence supporting independent causal roles for both Lp(a) and LDL on valve function. 16 The clinical implication, whether LDL-lowering has any role in AS beyond Lp(a) reduction, therefore remains unresolved.
Bicuspid Aortic Valve
The genetic and molecular drivers of AS vary significantly depending on the patient's underlying valve anatomy. 7 While BAV is a well-known risk factor for accelerated AS development, recent multiomics studies have begun to uncover the specific pathways that distinguish it from tricuspid aortic valve (TAV) disease.
A landmark 2026 meta-analysis by Thériault et al of over 9000 cases has established that BAV possesses a highly polygenic foundation. 21 The study identified 36 genomic loci, including 32 novel regions, implicating genes involved in heart morphogenesis such as WNT4 and LEF1. 21 Using transcriptomic data from human aortic valves, the researchers prioritized KANK2 and ERBB4 as potentially causal genes. 21 Their work suggests that the initial disruption of the valve is linked to common genetic variants that also increase the risk for long-term complications like thoracic aortic aneurysm. 21
The progression of AS in BAV patients is characterized by a unique molecular signature once calcification begins. Integrated transcriptomic and metabolomic analyses have revealed that BAV-associated AS is specifically characterized by heightened mitochondrial dysfunction and increased oxidative stress compared to TAV patients. 22
Transcriptomics: Decoding the Regulatory Networks of Valvular Disease
Transcriptomics offers a dynamic snapshot of the genes that are being expressed, revealing the cellular and regulatory processes that contribute to AS progression.
Bulk Transcriptomics and Key Signaling Pathways
While single-cell RNA sequencing (scRNA-seq) can reveal cellular heterogeneity and examine AS progression, bulk RNA-seq of larger patient cohorts remains crucial for validating pathway-level findings. The pathways and key genes are summarized in Table 2. Houessou et al conducted a transcriptome-wide profiling of human aortic valves to discover patterns in gene expression associated with AS severity, age of onset, immune cell composition, and valve morphology (TAV vs BAV). 23 In valves with more severe AS, genes related to immune response pathways were upregulated, whereas genes involved in lipid metabolism and peroxisome proliferator-activated receptors (PPAR) signaling were downregulated. 23 PPARγ, a member of the PPAR gene family, is a transcription factor involved in glucose homeostasis, adipocyte differentiation, and lipid metabolism. 23 Previous studies have also identified the downregulation of PPARγ in samples with AS, leading to inflammation. 23 Therefore, PPARγ agonists may serve as a potential therapeutic. Early age of AS onset demonstrated unique transcriptomic signatures depending on valve morphology. 23 In TAV, there was overexpression of IL6, IL1B, and IL24, whereas in BAV, there was downregulation of PPAR signaling genes. 23 Samples with AS also showed higher estimated proportions of B cells, neutrophils, macrophages (M0, M1), CD8 T cells, and NK cells, indicating an association between adaptive immune response and AS severity. Greene et al also conducted RNA-seq and found the key pathways related to AS were immune regulation and lipid homeostasis. 24 Compared to normal valves, valves with AS showed upregulation of COL10A1, COL11A1, HBB, IBSP, H19, and SPP1. 24
Key Genetic Loci Implicated in Aortic Stenosis.a

Abbreviations: SNP, single-nucleotide polymorphism; AS, aortic stenosis; BAV, bicuspid aortic valve; ASCVD, atherosclerotic cardiovascular disease; VIC, valvular interstitial cell; BMI, body mass index; ECM, extracellular matrix.
Loci are listed with their lead SNP, the biological pathway implicated by available evidence, and a summary of key findings. Functional validation has been demonstrated for a subset of these loci; see text for details.
Key Signaling Pathways and Genes Implicated in Aortic Stenosis.a

Abbreviations: AS, aortic stenosis; BMP, bone morphogenetic protein; MMP, Matrix Metalloproteinase.
Pathway-level findings from transcriptomic studies of human aortic valves. Direction of change refers to expression in AS valves compared with non-AS controls.
Key Regulatory Noncoding RNAs in Aortic Stenosis Pathogenesis.a

Abbreviations: AS, aortic stenosis; miRNA, microRNA; lncRNA, long noncoding RNA; EndoMT, endothelial-to-mesenchymal transition; LV, left ventricular.
Direction of change refers to expression in AS tissues compared with non-AS controls.
Protein and Metabolite Candidate Biomarkers Associated With Aortic Stenosis.a
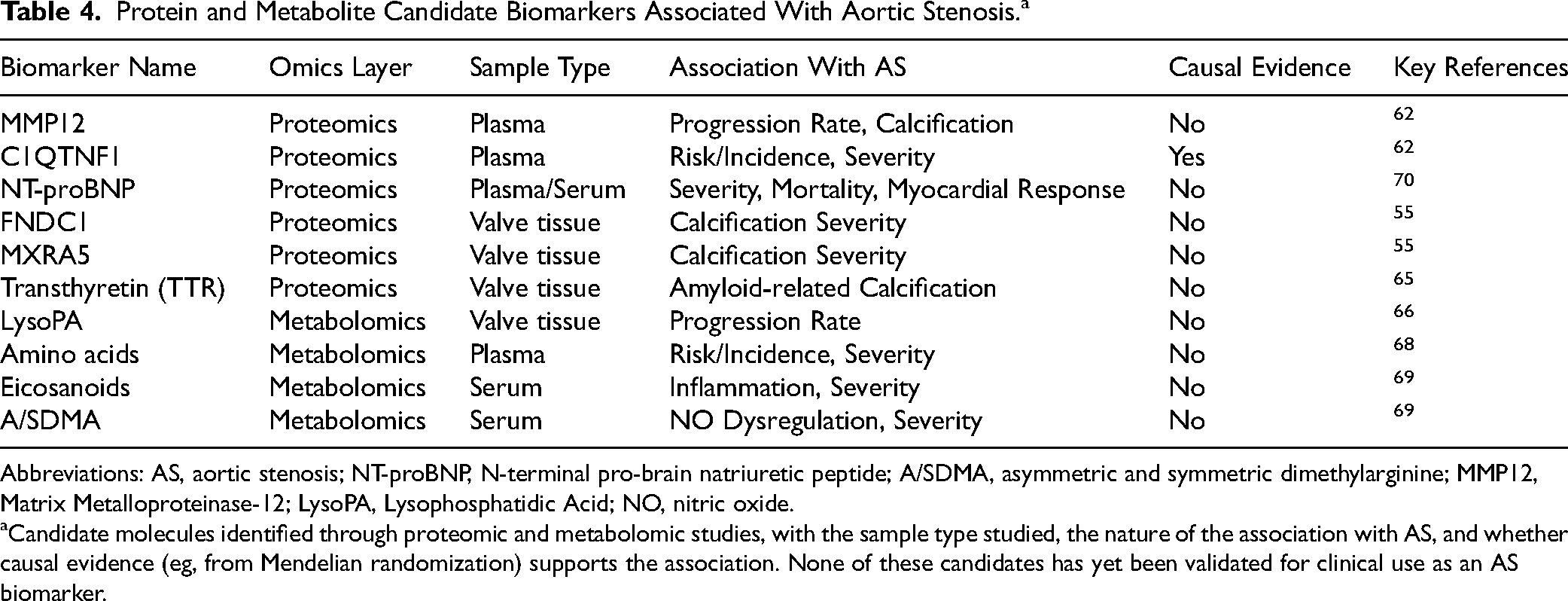
Abbreviations: AS, aortic stenosis; NT-proBNP, N-terminal pro-brain natriuretic peptide; A/SDMA, asymmetric and symmetric dimethylarginine; MMP12, Matrix Metalloproteinase-12; LysoPA, Lysophosphatidic Acid; NO, nitric oxide.
Candidate molecules identified through proteomic and metabolomic studies, with the sample type studied, the nature of the association with AS, and whether causal evidence (eg, from Mendelian randomization) supports the association. None of these candidates has yet been validated for clinical use as an AS biomarker.
Ossification, calcification, and fibrotic pathways are also involved in AS. In valves with severe AS, Houessou et al found upregulation of genes related to ossification and ECM organization, including IBSP, MMP9, and MMP13. 23 Guauque-Olarte et al performed RNA-seq to examine differences in gene expression between TAV and BAV with calcification. Both valve morphologies showed upregulation of fibrotic, immune response, and ossification pathways. 26 Gene expression was almost identical in end-stage AS in TAV and BAV. 26 There were multiple key signaling pathways that were enriched in calcified TAV and BAV: Bone morphogenetic protein (BMP) signaling (RUNX2 gene upregulated); Wnt signaling (RSPO2 gene upregulated); calcium signaling (CACN2D4, CACNA1C, and CACNA1H genes upregulated); and fibrosis signaling (MMP9, MMP11, MMP12, and MMP13 genes upregulated). 26
Semenova et al conducted a multiomics study taking a closer look at the osteogenic differentiation of valve interstitial cells (VICs), which contributes to aortic valve calcification. 27 The VICs isolated from 6 healthy valves and 6 valves with AS were compared after cultivation in standard conditions at 48 h after the induction of osteogenic differentiation (via osteogenic medium), and on day 10. 27 The initial phase (first 48 h) was characterized by the modulation of the Wnt, FoxO, and HIF-1 signaling pathways, indicating these pathways being involved in the commitment of VICs to osteogenic differentiation. 27 The later phase (day 10) was dominated by sustained activation of the PI3K-Akt, MAPK, and TNF signaling cascades. 27
Transcriptomic analyses have provided mechanistic insights into both risk factor-specific and disease-wide gene expression changes in AS. Bourgeois et al compared gene expression in calcified aortic valves with high versus low exposure to Lp(a), a known risk factor for the development of AS. 28 Patients with high Lp(a) levels showed a distinct transcriptomic signature from those with low Lp(a) levels. 28 Pathway analysis indicated that Lp(a) may be associated with dysregulation of genes involved in cellular aging, chondrocyte development, and inflammation, demonstrating a connection between this genetic risk factor and pathogenic gene expression, though no single gene met statistical significance. 28 Complementing this finding, Zamani et al leveraged RNA sequencing data from 500 human aortic valves to identify 100 protein-coding genes uniquely expressed in the aortic valve compared to 45 other tissues, 38 of which were differentially expressed in severe AS. 29 Ten of these genes showed a progressive expression gradient tracking with disease severity and were highly connected in a protein–protein interaction network. 29 The majority of these genes were involved in ECM regulation, including LUM, COL3A1, PRG4, and COMP, which increased with worsening disease, as well as TIMP3 and FBLN2, which decreased. 29 Inflammatory mediators such as C-X-C Motif Chemokine Ligand 12, TNFAIP6, and MIF were also prominently dysregulated, further supporting a role for inflammation in AS pathogenesis. 29
Single-Cell Transcriptomics
Single-cell RNA sequencing can capture cellular heterogeneity that bulk RNA sequencing may miss, and examining individual cells across different stages of AS progression may inform AS pathogenesis. 30 With scRNA-seq, researchers can examine layer-specific cellular processes (in the fibrosa, spongiosa, and ventricularis layers) and identify heterogeneity within subpopulations of the valvular cell types—VICs, valvular endothelial cells (VECs), and immune cells. It can also identify novel valve-derived stromal cell (VDSC) populations that appear only in the context of disease. 30 Xu et al found 6 novel VDSC clusters with scRNA-seq, marked by LUM, SOX4, CCL20, MT1A, RPL17, and CMSS1. 30
Xu et al used scRNA-seq to examine the relationship between endothelial-to-mesenchymal transition (EndoMT) and AS. 30 EndoMT refers to a phenotype change in endothelial cells involving a loss of endothelial characteristics and a gain of mesenchymal qualities. RNA sequencing confirmed an upregulation of mesenchymal markers and downregulation of endothelial markers in VECs from valves with CAVD. 30
Using scRNA-seq, Villa-Roel et al examined the layer-specific pathogenesis of AS. 31 The fibrosa experiences abnormal shear stress; therefore, it tends to undergo more AS-related changes than the ventricularis, which faces more regular, pulsatile forces. 31 ScRNA-seq showed that fibrosa-side VECs have a different transcriptional profile from ventricularis-side VECs, including greater upregulation of pro-inflammatory and profibrotic genes, as well as EndoMT markers. 31 They also found 49 side-dependent upregulated genes, such as members of the activator protein-1 (AP-1) transcription factor (FOSB, EGR1, JUN, and JUNB), especially in VECs of fibrosa with more severe AS.
Villa-Roel et al also examined cellular heterogeneity with different stages of AS. 31 They found VEC clusters changed with disease progression. Nondiseased aortic valve leaflets had more VEC1-3 clusters, which were mostly protective or homeostatic, whereas fibrotic or calcified leaflets had more VEC4-7 clusters, which were more pro-inflammatory and procalcific. 31 The VIC and immune cell clusters also changed with disease stage, shifting from homeostatic to inflammatory and fibrotic phenotypes with AS progression.5,31
Lyu et al focused on BAVs and examined macrophage involvement in disease progression, specifically macrophage-to-mesenchymal transition (MMT). 25 Using scRNA-seq, Lyu et al discovered a macrophage-derived stromal cell (MDSC) phenotype and evidence of MMT in BAVs with AS. The MDSCs originate from MRC1+ (CD206) macrophages and have both macrophage and mesenchymal markers. Lyu et al suggest that MRC1 + macrophages are involved in remodeling the ECM, and MDSCs are a source of osteoclast-like cells and myofibroblasts in stenotic aortic valves. 25
While single-cell sequencing has identified LUM as a hallmark of pathogenic VDSC clusters, recent functional work has clarified its role as a driver of the disease. Huang et al established that LUM promotes the transition of VICs into an intermediate inflammatory state before final osteogenic transformation. 32
Noncoding RNAs
A significant portion of the transcriptome consists of noncoding RNAs (ncRNAs), which do not code for proteins but instead, function as regulators of gene expression. In AS, microRNAs (miRs) and long ncRNAs (lncRNAs) have been investigated as molecular switches that influence the disease process (Table 3). 33
A systematic review synthesized studies that explored miRs related to AS. 34 However, results were inconsistent across studies, which highlights the need for further high-quality research in this area. 34 Key miRs identified in the systematic review include:
microRNA-21
The most frequently reported miR. microRNA-21 is found to be upregulated in both myocardial and valvular tissue with AS compared to control. 34 MiR-21 helps regulate the ERK-MAP kinase pathway, which is involved in the cardiac fibroblast response, and upregulation is associated with fibrosis.34–36 MiR-21 expression also changes with other diseases, including cancer.34,37
microRNA-133a
Generally found to be downregulated in valve tissue cases of severe AS. Upregulation of miR-133a was found to be associated with LV mass regression following AVR, suggesting it plays a protective role against hypertrophy and fibrosis.34,38–41
Other Key miRNAs
In myocardial tissue, miR-19b was downregulated in AS patients relative to controls, while findings for miR-1 were inconsistent.34,42,43 In aortic valve tissue, miR-665, miR-602, and miR-939 were downregulated in stenotic valves relative to nonstenotic control valves, whereas miR-193b and miR-214 were upregulated.34,44–48
Long ncRNAs are another type of ncRNA with regulatory functions. Several lncRNAs have been identified as key players in AS:
H19
Emerged as a central pro-osteogenic regulator. In AS, the promoter region of the H19 gene becomes hypomethylated, which leads to overexpression of H19 in the valve. 46 ,47 Overexpressed H19 induces an osteogenic phenotype by interfering with the antiosteogenic NOTCH1 pathway. A downstream target of NOTCH1, HEY1, is a repressor of BMP2 and RUNX2, which contribute to osteogenesis. H19 prevents the transcription factor p53 from binding to the NOTCH1 promoter, which decreases expression of HEY1, thereby lifting the brakes on calcification. 46 H19 may also play a role in promoting EndoMT, a mechanism that contributes to AS pathogenesis. 48
WISPER
Regulates cardiac fibrosis and is enriched in cardiac fibroblasts. WISPER expression was upregulated in cardiac tissues with AS.48,49
Chast
Expression is induced in hypertrophic heart tissue from AS patients. Chast helps regulate autophagy, and overexpression may promote cardiac hypertrophy.48,50
Sex-Specific Transcriptomics
Lesions in AS present differently in females compared to males: for a similar degree of severity, women with AS often present with more valvular fibrosis, whereas men tend to have more extensive calcification. 51 Transcriptomic analysis of stenotic valves has helped provide a molecular basis for these sex-specific phenotypes by identifying genes that are differentially regulated between men and women with AS. 51 Le Nezet et al found that in women, there was an overexpression of genes involved in fibrosis, such as TGFβ2, KIF1A, and FRAS1, as well as genes related to apoptosis, such as SFRP4, CES4 and TGFB2. 51 Both males and females had upregulated genes associated with calcification: in female patients, TPD52L1 and RCN2 were upregulated, whereas men showed a relative upregulation of procalcific genes STC2 and CPAMD8, along with the MAPK pathway. 51 These distinct transcriptional profiles may be partially explained by sex chromosome-linked epigenetic mechanisms. Gorashi et al demonstrated that UTY, a Y chromosome-linked lysine demethylase, uniquely decreases methylation in male VICs in response to nanoscale ECM cues. 52 This change promotes an osteoblast-like phenotype that drives the predominant calcification observed in males. 52 In contrast, female VICs, which lack the Y chromosome, more readily activate as myofibroblasts, contributing to the fibrotic phenotype characteristic of female AS. 52 These findings highlight the importance of sex as a biological variable in future therapeutic development.
Proteomics: Identifying Functional Effectors and Candidate Circulating Biomarkers
Compared with the rapidly expanding GWAS and transcriptomic evidence base, proteomic studies of AS remain fewer in number and smaller in scale, and the field is at an earlier stage of consolidation. Within this evolving landscape, proteomic analyses of both valve tissue and blood plasma have identified candidate effector molecules driving pathological remodeling and circulating proteins associated with AS that may eventually be developed as noninvasive markers or therapeutic targets.
Tissue Proteomics
Analysis of the proteome of surgically explanted aortic valves provides a view of the molecular composition of the diseased tissue. A consistent and central finding from studies that use tissue samples is a disruption of the ECM proteome.55,56 In calcified valves, there is an overexpression of procalcifying ECM proteins such as fibronectin (FN1) and osteopontin (SPP1), alongside a downregulation of key metabolic enzymes, such as ALDH1, ENO1, and SOD1, which indicates a link between structural remodeling and metabolic dysfunction. 55 In addition, Bouchareb et al identified novel ECM-related biomarkers whose expression levels in valvular tissue correlate directly with the severity of calcification. 55 Two of these biomarkers are FNDC1, which plays a role in matrix calcification with SPP1, and MXRA5, the role of which in AS needs further research. 55 Xu et al identified 6 proteins expressed in aortic valve tissue that demonstrate a relationship with AS: ANGPTL4, PCSK9, ITGAV, CTSB, GNPTG, and FURIN. 57 These proteins are involved with calcification, lipid regulation, blood pressure, and fibrosis. 57 Khan et al found a higher abundance of Wnt pathway mediators, GSK-3β, DVL2, β-catenin, and SFRP2, in human valves with AS compared to healthy valves, consistent with transcriptomic findings discussed above. 58 In human valves resected during surgery, Han et al found that calcified areas, compared to noncalcified areas, had higher expression of SPP1 and osteonectin (SPARC). 59 SIRT2, a deacetylase involved with stress response, was also found with higher levels in calcified valvular tissue compared to healthy tissue and may increase calcification under stress. 60 Differential electrophoresis and mass spectrometry performed on valve samples with and without AS identified several proteins that are increased, such as apolipoprotein A-I, lumican, vimentin, as well as proteins that are decreased, including transgelin, HSP27, in stenotic valves. 61 The proteins identified in this study play a role in a wide range of biological processes, including fibrosis, homeostasis, and coagulation, but further research is necessary to better understand these processes and their relation to AS. 61
Plasma Proteomics: Candidate Circulating Biomarkers
In addition to tissue analysis, studying proteins in the plasma is important in the discovery of biomarkers. Large-scale plasma proteomics has created an opportunity to survey thousands of proteins with potential causal relationships with AS. 62 Studies have identified dozens of circulating proteins associated with AS severity, progression, and incident clinical events (Table 4). These studies surveyed the circulating proteome using affinity-based platforms—aptamer-based assays (SomaScan) and antibody-based proximity extension assays (Olink). Shelbaya et al measured ∼4900 proteins by SomaScan, Bortnick et al used the SomaLogic platform of ∼5000 proteins, and Tan et al profiled 183 proteins by Olink. Key proteins include:
Matrix Metalloproteinase-12
Elevated circulating levels of Matrix Metalloproteinase-12 (MMP12) are associated with worse AV hemodynamic progression (measured by an increase in AV peak velocity) and greater AV calcification on CT imaging. 62 The MMP12 is also associated with atherosclerosis and aortic aneurysm and should be regarded as a candidate marker rather than a disease-specific one.
Complement C1q TNF-Related Protein 1
Another candidate biomarker is associated with worse AV hemodynamics. Mendelian randomization analyses suggested that genetically determined levels of Complement C1q TNF-Related Protein 1 (C1QTNF1) may have a causal effect on AS development. 62
Matrix Metalloproteinase-1
A protein involved in matrix remodeling. When overexpressed, MMP1 likely promotes calcification via ECM degradation and signals inflammation. 60
Leptin
A protein involved in metabolic processes. Higher circulating leptin was found to be associated with less severe AS and a lower risk of hospitalization; in AV tissue transcriptomics, leptin expression was higher in fibrotic compared with normal AV segments, but did not differ between calcific and fibrotic tissue. 62
KLKB1 (Kallikrein)
Bortnick et al found that higher levels of plasma KLKB1 were associated with lower levels of aortic valve calcification. Mendelian randomization suggested a causal protective role, meaning KLKB1 might actively help prevent the calcification process. 63
C-X-C Motif Chemokine Ligand 12
An inflammatory protein is positively associated with aortic valve calcification. It is a novel discovery in the context of aortic valve disease, highlighting the role of specific inflammatory pathways in valve remodeling. 63
Sex-Specific Proteomics
Matilla et al examined the sex-specific role of a particular protein, Gal-3, in AS progression. 64 Using VICs isolated from AVs with severe AS that were removed during AVR, they found Gal-3 expression was significantly higher in males than females with the same AS severity. In men, Gal-3 was primarily expressed by activated VICs and endothelial cells, whereas in women, Gal-3 was expressed by quiescent cells. 64 The findings of this study suggest that Gal-3 contributes to differences in AS progression between men and women and demonstrates the importance of considering sex-specific proteomics of AS when developing therapeutics.
The Amyloid-Stenosis Hypothesis
There is a link between AS and amyloidogenesis. 65 Heuschkel et al found a molecular network in CAVD that showed an overlap with pathways implicated in Alzheimer's disease. 65 Immunohistochemistry confirmed the physical presence of 2 amyloidogenic proteins-transthyretin and amyloid precursor protein—in the calcified regions of human aortic valves. 65 This discovery suggests that the processes of protein misfolding and aggregation, commonly studied in the context of neurodegenerative diseases, may be important pathogenic drivers in AS.
Validation, Clinical Applicability, and Limitations
Validation of candidate plasma proteins across cohorts and platforms remains early-stage in AS. The largest studies to date, Shelbaya et al, Bortnick et al, and Tan et al, used different assay platforms and different patient populations. Although they identify partially overlapping protein signatures, no single candidate has yet been validated to the standard required for clinical use. Cross-platform concordance has been demonstrated for selected proteins: in the Shelbaya et al analysis, MMP12 measurements by the SomaScan aptamer assay and the Olink proximity extension assay correlated at r = 0.91, supporting analytical robustness. Whether this technical concordance translates to clinical-grade reproducibility across diverse populations has not been tested.
Clinical applicability of these candidate proteins is constrained by several factors. None of the leading candidates is specific to AS; MMP12 is also associated with atherosclerosis and aortic aneurysm. Most other identified proteins reflect broader cardiovascular biology rather than valve-specific processes. The effect sizes observed in association studies are modest, raising questions about whether a single-protein test could meaningfully add to existing clinical risk stratification. Assay development for clinical use would also require a transition from research platforms (SomaScan, Olink) to standardized clinical immunoassays with established reference ranges, a step that has not been taken for any of the candidate AS proteins. The most realistic near-term clinical applications are likely to be in risk stratification or progression monitoring of patients with established mild-to-moderate AS, rather than population-level screening.
The current proteomic evidence base also has structural limitations. Most studies are cross-sectional, limiting inference about temporal dynamics; sample sizes for incident AS endpoints remain modest by GWAS standards; the major cohorts (ARIC, CHS) are predominantly of European ancestry; and the affinity-based platforms, while highly multiplexed, may miss proteins poorly represented on the assays. Mendelian randomization has identified C1QTNF1 as a candidate causal protein, but the broader question of which associated proteins are causal drivers versus markers of downstream pathology remains largely unresolved.
Metabolomics
Metabolites directly reflect the molecular phenotype, integrating genetic, epigenetic, and environmental factors to identify the metabolic interferences driving the disease.
Untargeted metabolomic profiling of explanted human aortic valves has enabled the creation of a “metabolite atlas,” identifying dozens of metabolites that are significantly altered across the spectrum of disease severity, from mild sclerosis to severe calcification. 66 Pathway analysis of these altered metabolites consistently reveals that the most affected metabolic pathways in AS involve lipid metabolism, particularly glycerophospholipid metabolism and bile acid biosynthesis. 66
Key Metabolites
Several specific metabolites have been identified as key contributors to AS pathogenesis and progression:
Lysophosphatidic Acid
Tissue levels of Lysophosphatidic Acid (LysoPA) species correlate positively with the rate of hemodynamic progression of AS. 66 The LysoPA is a potent stimulator of VIC inflammation and osteogenic differentiation, generated when autotaxin acts on the oxidized phospholipids carried by Lp(a). 67 The full cascade, from the LPA locus through Lp(a), autotaxin, and LysoPA to VIC calcification, is the cohesive multiomics pathway summarized in the integration section below and depicted in Figure 1B.
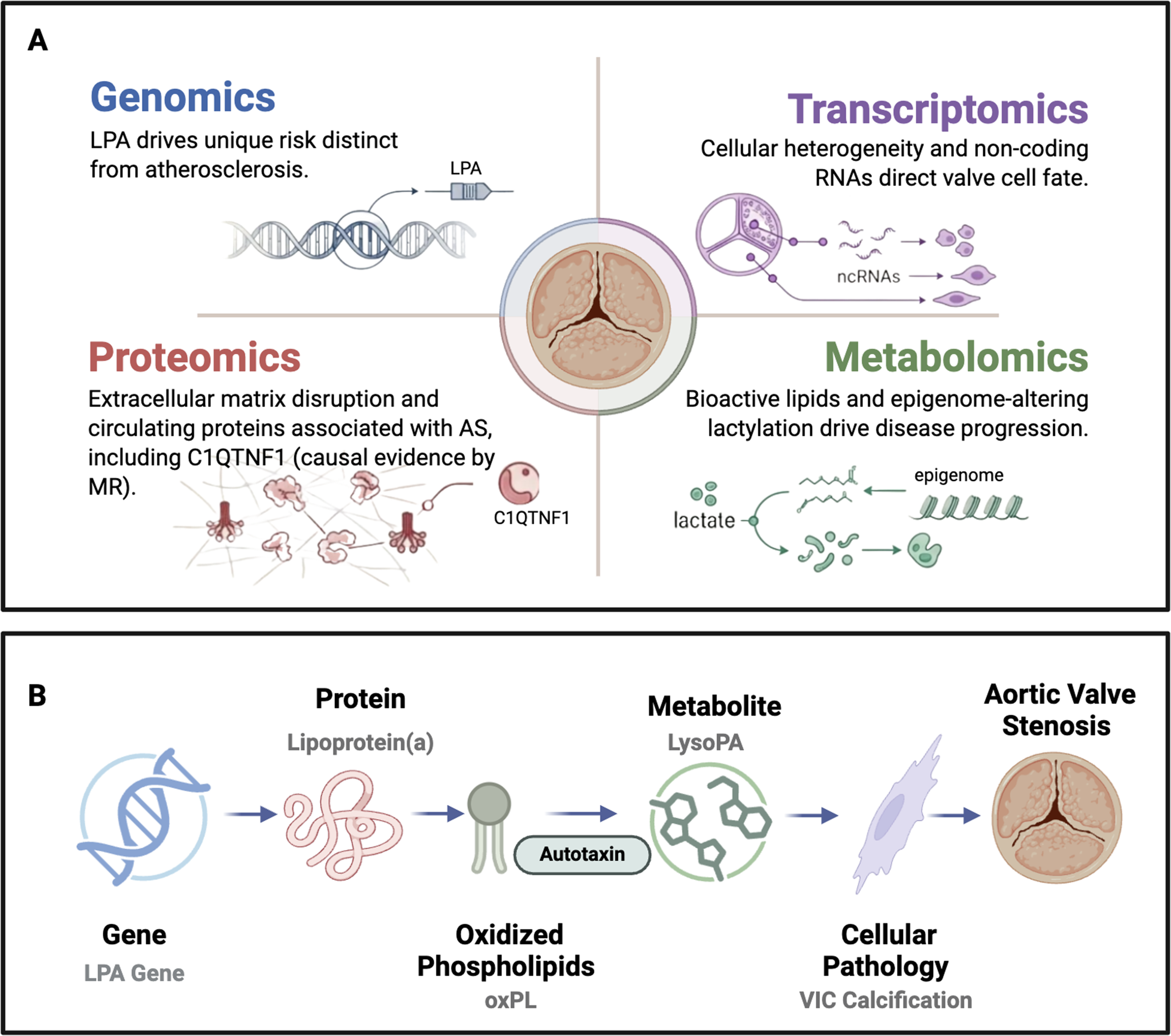
Multiomics characterization of aortic stenosis. (A) Each omics domain contributes a distinct layer of evidence: genomics identifies inherited risk loci (most notably LPA) with a genetic architecture distinct from atherosclerosis; transcriptomics reveals cellular heterogeneity and regulatory noncoding RNAs that direct valve cell fate; proteomics identifies extracellular matrix disruptors and circulating proteins associated with aortic stenosis (AS), including Complement C1q TNF-Related Protein 1 (C1QTNF1), for which Mendelian randomization supports a causal role; and metabolomics maps bioactive lipid signaling and epigenome-altering lactylation as drivers of disease progression. (B) These layers converge on cohesive pathogenic axes. The Lp(a)-autotaxin-LysoPA cascade is a key example. Variation at the LPA locus (genomics) determines circulating lipoprotein(a) levels (proteomics); Lp(a) carries oxidized phospholipids (oxPL), which the enzyme autotaxin converts to lysophosphatidic acid (LysoPA, metabolomics); LysoPA stimulates valvular interstitial cell osteogenic differentiation and calcification (transcriptomics); culminating in aortic valve stenosis. Icon colors indicate the corresponding omics layer: blue, genomics; pink, proteomics; green, metabolomics; purple, transcriptomics.
Markers of Bone Turnover
The link between valvular calcification and bone biology is further strengthened by metabolomic findings. The detection of pyridinoline and deoxypyridinoline—well-established markers of collagen breakdown and bone turnover—in stenotic valve tissue provides direct chemical evidence for the osteogenic-like processes occurring within the valve. 66
Amino Acids and Fatty Acids
Large, community-based cohort studies such as ARIC have extended these findings to the circulating metabolome. These studies have demonstrated that elevated levels of certain circulating amino acids and amino sugar metabolites are predictive of both greater hemodynamic stenosis and a higher risk of future AS-related hospitalizations. Conversely, higher levels of dicarboxylic fatty acids appear to be protective, associated with better valve hemodynamics and a lower risk of clinical events. 68
LUM–Metabolome–Epigenome Cascade
Huang et al demonstrated that Lumican (LUM)-induced glycolysis leads to H3 histone lactylation (specifically H3K14la and H3K9la). 32 This process directly facilitates the transcription of osteogenic genes such as Runx2 and BMP2, representing a novel LUM-metabolome-epigenome cascade in valve calcification.
Inflammation and Oxidative Stress
Metabolomic profiling of serum from patients with severe AS reveals a dominant signature of inflammation and dysregulated nitric oxide (NO) metabolism.
69
In one study, 17 of the top 30 metabolites that distinguished AS patients from healthy controls were related to NO synthesis. This included increased levels of NO substrates (eg,
Metabolic Differences in BAV Versus TAV
Metabolomics also reveals distinct molecular features in AS associated with BAVs. An integrated study combining transcriptomics and metabolomics demonstrated that AS in BAV patients is characterized by a unique metabolic profile linked to mitochondrial dysfunction and heightened oxidative stress when compared to AS in patients with TAV. 22
Validation, Clinical Applicability, and Limitations of Metabolomic Evidence
Cross-cohort validation of metabolomic findings in AS is even less mature than for proteomics. The major tissue metabolomic study by Surendran et al was performed in a single cohort of explanted valves, and the ARIC plasma metabolomic findings have not yet been independently replicated. 66 Untargeted metabolomic profiling is inherently exploratory: it generates hypotheses about pathway involvement but typically yields modest effect sizes for individual metabolites and depends heavily on annotation databases that remain incomplete, particularly for lipid species.
Clinical applicability of metabolomic findings is constrained by the same translation challenges as proteomics, with additional sample-handling sensitivities. Many lipid metabolites, including the LysoPA species central to the autotaxin axis, are sensitive to dietary state, sample collection protocols, and freezer storage time, complicating standardization across clinical sites. Metabolomic measurements also do not yet have established reference ranges for AS-relevant populations. The most direct clinical translation of the metabolomic findings to date is not as a diagnostic test but as therapeutic-target identification: the autotaxin–LysoPA axis emerged from metabolomic evidence and is now in preclinical drug development, illustrating how metabolomics can inform therapeutic strategy even when individual metabolites are not yet clinically actionable as biomarkers.
Limitations specific to the AS metabolomic literature include the predominance of tissue-based studies in patients with severe disease undergoing AVR (which biases findings toward end-stage biology), the small number of large prospective cohorts with stored samples and longitudinal AS phenotyping, and the limited methodological harmonization across studies. These constraints argue for caution in interpreting individual metabolite findings and for prioritization of pathway-level signals (such as the consistent finding of lipid metabolism disruption) over single-metabolite biomarker claims.
An Integrated, Systems-Level View and Future Directions
The multiomics approach allows researchers to develop comprehensive, systems-level models of AS pathogenesis. This integration has revealed several interconnected pathogenic pathways, most notably those linking lipid metabolism, inflammation, fibrosis, and calcification.
Informing Clinical Trials
The ultimate purpose of this research is the development of pharmacotherapies that halt or reverse AS progression. For example, the strong causal evidence for Lp(a) has inspired the clinical development of Lp(a)-lowering therapies, such as antisense oligonucleotides and small-interfering RNAs. While these therapies are primarily under investigation for ASCVD, their potential effect on AS is also being explored. 8
Ongoing Trials
Lp(a) has entered Phase 3 testing as a target. Pelacarsen, an antisense oligonucleotide that lowers apolipoprotein(a) by approximately 80%, 71 is the subject of the Lp(a)HORIZON cardiovascular outcomes trial (NCT04023552) in patients with established ASCVD and elevated Lp(a). 71 Two large siRNA programs are also in late-stage development: olpasiran in the OCEAN(a) trial (NCT05581303), 72 and lepodisiran in the ACCLAIM-Lp(a) trial (NCT06292013). 73 The trial of greatest direct relevance to AS is Lp(a)FRONTIERS CAVS (NCT05646381), a Phase 2 trial of pelacarsen specifically powered to assess progression of calcific AS in patients with elevated Lp(a), currently recruiting with primary completion estimated in 2030. 71
Translation remains preclinical for other multiomics targets. Autotaxin inhibition has shown efficacy in valvular interstitial cell and animal models. BBT-877 is an orally available inhibitor that attenuates VIC osteogenic differentiation and reduces valvular calcification in murine and rabbit fibrocalcific aortic valve disease models. 74 However, no human AS trial has yet been initiated.
Feasibility Considerations
The feasibility of these therapies for AS depends on 3 biological realities. First, the Lp(a)-lowering trials are powered primarily for atherosclerotic endpoints, and even strongly positive outcomes in the ASCVD trials do not guarantee AS benefit. Second, the AS multiomics literature consistently points to end-stage tissue as the source of most molecular data, meaning many candidate targets reflect late disease biology; effective pharmacotherapy will likely require treating patients earlier in the disease course, before irreversible calcification. This has implications for patient identification and trial design. Third, the multipathway architecture of AS suggests that single-target therapies may produce modest effects on hemodynamic progression even if mechanistically correct.
Barriers to Implementation
Beyond the trials themselves, several practical barriers must be addressed before multiomics-derived therapies enter routine AS care. Disease identification is the most immediate: most patients are currently diagnosed only after symptoms develop. At this point, intervention with pharmacotherapy is unlikely to reverse established calcification. Imaging-genomic approaches such as the deep-learning-derived CMR phenotyping reported by Kany et al 16 may eventually enable identification of high-risk patients decades earlier, but this paradigm has not been validated in prospective clinical workflows. Drug delivery, cost, and ancestry-equitable access also present barriers. Lp(a)-lowering therapies will be expensive, require parenteral administration (with the exception of the oral inhibitor muvalaplin), and have been developed almost entirely in cohorts of European ancestry, mirroring the limitations of the underlying multiomics evidence base. Finally, even if pharmacotherapy proves effective at slowing progression, integration with existing AVR-based care pathways will require new clinical decision frameworks for when to treat medically, when to monitor, and when to proceed to valve replacement.
Challenges and the Future of Multiomics in AS
Despite progress, the multiomics approach faces significant challenges that must be addressed to realize its full potential in treating AS.
Protocols must be standardized for sample collection, processing, and data analysis to ensure that results are comparable and reproducible across different research groups and cohorts. 5 A major limitation is that the vast majority of AS research has been conducted in cohorts of European ancestry. Expanding studies to include more diverse populations is critical for understanding the full genetic and molecular spectrum of the disease and ensuring that future therapies are equitable. 5 Another significant challenge is the reliance on tissue samples obtained at the time of AVR. This means that most of our molecular data comes from end-stage disease, leaving our understanding of the crucial early initiating events of CAVD incomplete. 5 Furthermore, it is difficult to separate molecular signals originating from the primary valve pathology from those reflecting the secondary myocardial response to pressure overload. Many circulating biomarkers, such as NT-proBNP and troponins, are markers of cardiac stress and damage, not direct measures of valvular disease. 6 Therefore, future studies must integrate multiomics data with clinical phenotyping, especially advanced cardiac imaging techniques such as CMR, to accurately attribute molecular signatures to either the “valve disease” or the “myocardial response” component of the overall syndrome. 70
Reproducibility across studies remains a concern: differential expression findings have varied between cohorts, single-cell taxonomies of the diseased valve have not yet converged on a unified cell-population framework, and individual transcriptomic studies have often been underpowered to detect modest effects at the single-gene level. For example, the Bourgeois et al analysis of Lp(a)-stratified valves identified a distinct transcriptomic signature at the pathway level but found no individual gene meeting statistical significance. 28
Across all omics layers, the AS multiomics literature remains in a phase of rapid expansion, with the largest GWAS and proteomic studies published only within the past 3 years. Findings should therefore be interpreted with the expectation that the field will continue to evolve and consolidate as cohorts grow, methods mature, and replication studies accumulate.
Future Directions
The future of AS research should focus on overcoming these challenges and employing next-generation technologies.
Single-Nucleus Multiomics
Single-nucleus RNA sequencing offers specific advantages over single-cell sequencing for valve tissue. Calcified valves are fibrotic and resist the enzymatic dissociation that single-cell methods require, which biases single-cell data toward cells that survive the process and can introduce dissociation artifacts. Single-nucleus methods work on frozen, archived tissue and recover fragile or large cell types that single-cell suspensions miss, capturing the valve's cellular repertoire more completely. Coupling nuclear transcriptomics with parallel measurement of other omics layers in the same nucleus would link gene regulation directly to cellular function within specific pathogenic subtypes.
Artificial Intelligence and Machine Learning
Artificial intelligence (AI) and machine learning algorithms may help in identifying complex, nonlinear patterns, building predictive models for patient risk stratification, and personalizing treatment decisions. 75 Kany et al demonstrated the feasibility of “imaging-genomics,” where AI-derived phenotypes from routine CMR scans can reveal the genetic basis of subclinical valve remodeling. 16 This allows for the identification of high-risk patients decades before they develop severe stenosis, shifting the clinical focus from end-stage intervention to early-stage prevention.
Longitudinal Cohort Studies
There is a need for large-scale, prospective, longitudinal cohort studies that collect serial multiomics data and imaging from individuals over many years to understand the progression of the disease. These studies allow researchers to map the trajectory of CAVD from onset to severe stenosis, identifying critical transition points and planning precisely timed therapeutic interventions.
Conclusion
Multiomics has revolutionized our understanding of calcific AS. Genomics has helped establish a distinct genetic identity for AS and highlighted key causal drivers like lipoprotein(a). Transcriptomics, especially at single-cell resolution, has revealed the specific cell populations and regulatory networks, including ncRNAs, which orchestrate fibrosis and calcification. Proteomics and metabolomics have identified the functional protein effectors and metabolic drivers of the disease.
The power of this approach lies in integration. A systems-level view has provided clear molecular explanations for long-standing clinical observations, such as the failure of atherosclerosis-targeted therapies and the sex-specific differences in disease presentation. The Lp(a)-autotaxin-LysoPA pathway is a key illustration of how this model can connect source to pathophysiology. While significant challenges remain, particularly in studying early-stage disease and ensuring population diversity, continued application of multiomics can guide the development of targeted pharmacotherapies and precision diagnostics.
Footnotes
Acknowledgments
During the preparation of this work, the authors used Google Gemini Deep Research to assist with the initial identification of relevant literature across the genomics, transcriptomics, proteomics, and metabolomics domains and to help organize the preliminary structure of the review. All references identified through this process were independently screened and evaluated by the authors. References deemed unsuitable were excluded, and additional primary literature sources were identified and incorporated through independent literature searches conducted by the authors. All references included in the final manuscript were reviewed directly from the original publications.
ORCID iDs
Ethics Approval
Not applicable.
Author Contributions
Nora Treleaven contributed to writing—original draft, review & editing. Sarah Gillies contributed to writing—original draft. Pardis Zamani contributed to writing—review & editing. Anne-Marie Seguin contributed to writing—review & editing. Yohan Bossé contributed to writing—review & editing. Sébastien Thériault contributed to writing—review & editing. Guillaume Paré contributed to writing—review & editing, supervision, and conceptualization. Richard Whitlock contributed to writing—review & editing, supervision, and conceptualization.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability
This review article does not contain original data. All data discussed are available in the primary studies cited within the article.