
Abstract
Lower urinary tract obstruction (LUTO), anorectal atresia, exomphalos, congenital diaphragmatic hernia (CDH), and disorder of sexual differentiation (DSD) are severe congenital abnormalities. Prenatal diagnosis of each abnormality has been described in early pregnancy with varying consistency. This is likely the first published report of a sporadic presentation of all five malformations combined identified in a single early pregnancy case. A 31-year-old primigravida woman was referred for a subsequent sonogram following identification of dilated large bowel loops on a previous sonogram completed at 13 weeks 3 days, gestational age. Further sonographic assessment identified exomphalos and a distended urinary bladder. The postmortem examination done at 15 weeks 6 days gestation confirmed these defects, and further identified CDH and a DSD. This set of fetal anomalies does not fit the diagnostic criteria of known syndromes, sequences, complexes, or associations.
Keywords
Lower urinary tract obstruction (LUTO), anorectal atresia, exomphalos (or omphalocele), congenital diaphragmatic hernia (CDH), and disorder of sexual differentiation (DSD) are fetal abnormalities with high rates of morbidity and mortality. The occurrence of LUTO (incidence 3.34 per 10 000 births) 1 is often caused by posterior urethral valves or urethral atresia. It is classically identified by the triad of megacystis, posterior urethral valve (“keyhole sign”), and hydronephrosis. Complications from LUTO mainly arise from oligohydramnios, which is secondary to reduced fetal urine output, causing pulmonary hypoplasia and limb defects.2 –4 Anorectal atresia (anorectal malformation, anal atresia, anal malformation, anal/anorectal agenesis) is often syndromic and associated with other congenital fetal malformations (e.g., VACTERL association), with a total incidence of 3.26 per 10 000 births. 5 There is some evidence for postnatal surgical intervention of anorectal atresia in isolation, although the presence of other malformations significantly impacts prognosis.6,7
Exomphalos is an abdominal wall defect (incidence 3.8 per 10 000 births) 8 causing midline herniation of intra-abdominal viscera through the base of the umbilical cord contained within the parietal peritoneum. Like anorectal atresia, it has a high association and worsening prognosis when present alongside other congenital abnormalities.9 –11 The diagnostic differential may include gastroschisis, whereby intra-abdominal contents herniate feely without a peritoneal sac. Gastroschisis is not associated with other abnormalities and has a significantly better prognosis than an omphalocele. Differentiation between the two is usually achieved by the absence of a peritoneal sac and normal umbilical cord insertion into the abdominal wall in gastroschisis compared with omphalocele. 9
Congenital diaphragmatic hernia (incidence 2.3 per 10 000 births) 12 describes the consequences of a weakness in the fetal diaphragm, causing herniation of intra-abdominal contents into the thorax which reduces capacity for lung development, leading to pulmonary hypoplasia and pulmonary hypertension. Congenital diaphragmatic hernia may be suitable for in utero or postnatal intervention, with variable prognosis depending on the organs involved and laterality of herniation.10,13,14 The primary differential diagnosis for CDH is congenital cystic adenomatoid malformation (CCAM), a rare congenital cystic malformation syndrome with significantly better outcomes than CDH. 15 Differentiation from CCAM on a prenatal sonogram includes visualization of a diaphragmatic defect, stomach localization within the chest, and lack of cystic areas within pathological intrathoracic lesions. 16 Congenital diaphragmatic hernia, unlike CCAM, is generally associated with chromosomal abnormalities and other congenital abnormalities. 17
The term DSD encompasses a range of phenotypes not following normal sexual development, which are usually chromosomal, gonadal or anatomical in origin. 18 Disorder of sexual differentiation is a broad term that covers any presentation of atypical genitalia, replacing the obsolete term hermaphroditism. 19 Severity may range from simple anatomical malpresentation compared with expected binary male or female genitalia, treatable with surgical intervention, to infertility and salt wasting crises depending on etiology. 20 The prenatal detection rate of DSDs is low, possibly 15% to 24%, and early presentation in pregnancy may be associated with more severe phenotypes.21,22 If present during sonographic screening, the main typifying feature is ambiguous genitalia.22,23 Disorder of sexual differentiation has an unclear incidence due to an ambiguity over classification, but is estimated to be present in between 1 in 4500 and 1 in 5000 live births, with the most common of these 46,XY DSD due to congenital adrenal hyperplasia.24 –26
The first trimester presentation of LUTO is well documented within the literature and early pregnancy anorectal atresia has been reported previously. However, there have been no accounts of these abnormalities occurring alongside CDH, exomphalos and a DSD simultaneously in early pregnancy. This is likely the first reported case, based on the available literature, of an early pregnancy afflicted with the combination of LUTO, anorectal atresia, exomphalos, DSD, and CDH.
Case Report
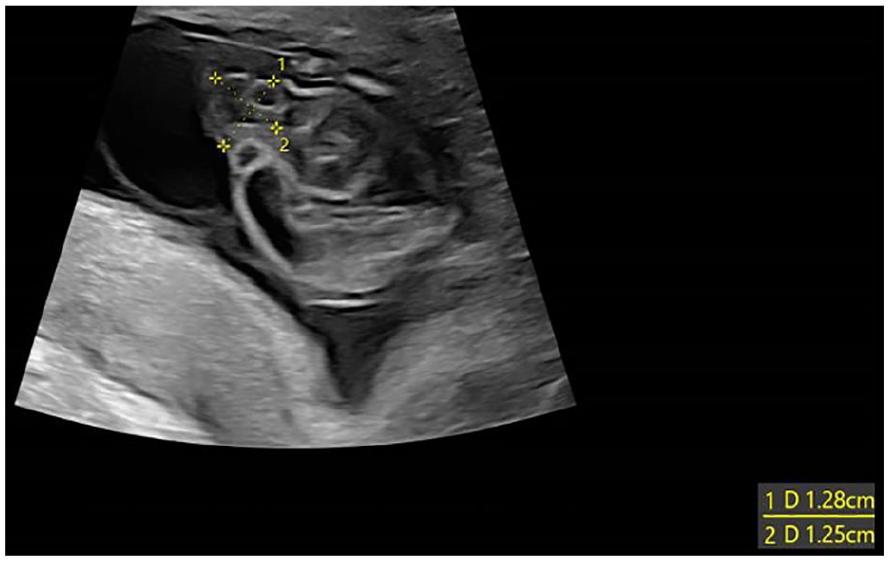
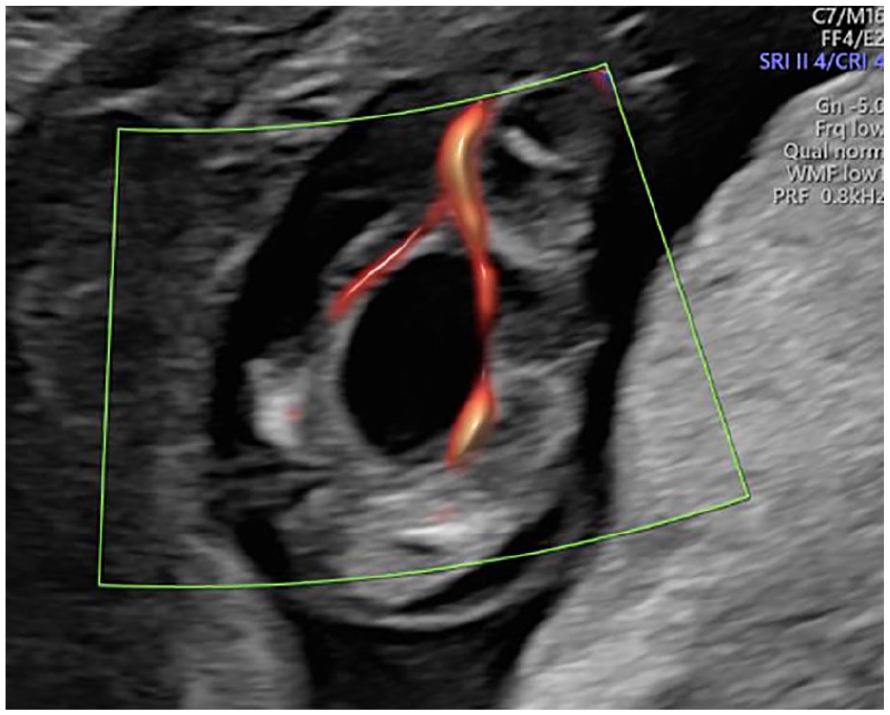
A 31-year-old primigravida woman was referred for a subsequent sonogram following the identification of dilated large bowel loops on prior fetal sonographic study (See Figure 1). The transabdominal sonogram was performed with an empty maternal bladder, using a Voluson E10 (GE Healthcare, Waukesha, Wisconsin) ultrasound equipment system and an RM6C convex transducer (2.0-6.0 MHz bandwidth). This sonographic examination was completed at 13 weeks 3 days. This sonographic study confirmed multiple dilated large bowel loops, increased abdominal circumference, and herniation through the umbilical aperture, which is consistent with exomphalos (See Figure 2). Further sonographic findings identified a large cystic lesion in the lower part of the fetal abdomen, believed to be a distended urinary bladder (See Figure 3). Both kidneys were visualized with good vascular flow (See Figure 4), while the fetal chest imaging demonstrated good cardiac biventricular flow with normally sited lungs (See Figure 5).
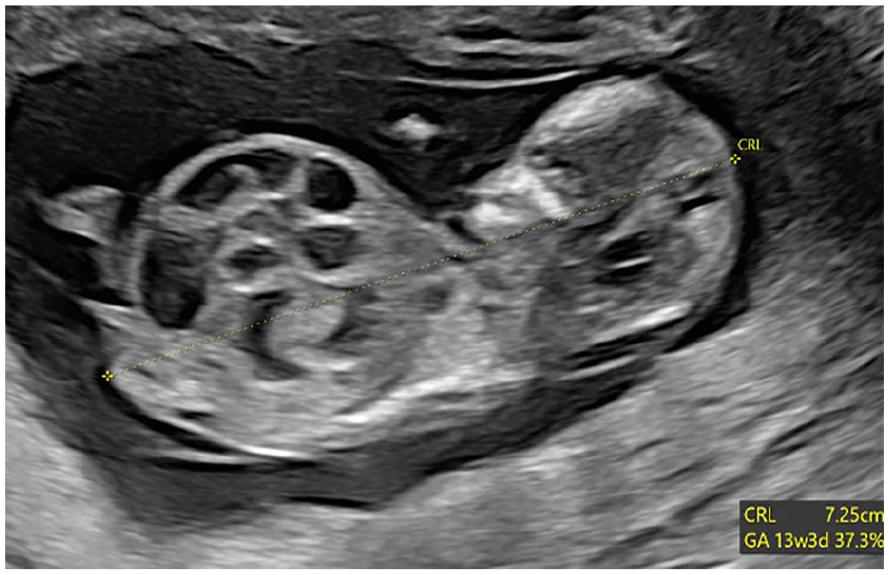
This sonographic sagittal view of the fetus demonstrates multiple distended bowel loops.
A sonographic axial view of the fetus demonstrates an omphalocele containing dilated bowel loops.
This sonographic axial view of the fetal pelvis demonstrated a distended urinary bladder flanked by umbilical arteries.
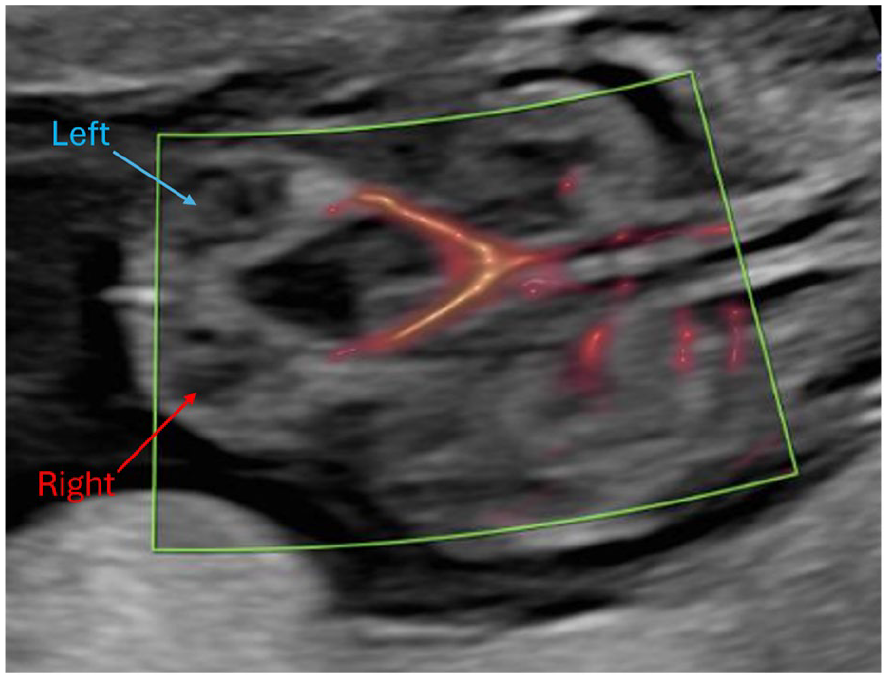
This sonographic coronal view of the fetus shows both kidneys (blue = left kidney, red = right kidney) and the associated vasculature.
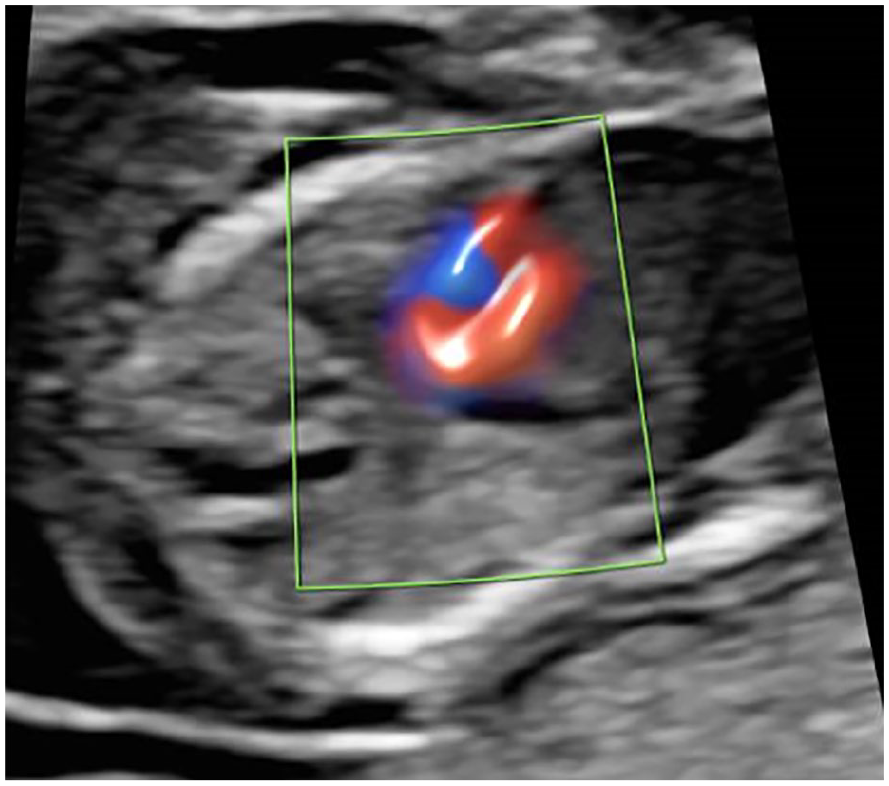
A sonographic axial view of the fetal chest demonstrates cardiac biventricular color flow and normally sited lung tissue.
A viral infection and cystic fibrosis screening were performed, both of which had a negative diagnostic result. A posterior, high placenta prevented chorionic villus sampling, and amniocentesis was unavailable until 16 weeks, which prevented early chromosomal and genetic testing. The family ultimately opted for a termination of this pregnancy, which was performed at 15 weeks 6 days. This termination was done using a Mifepristone-Misoprostol induction regime as was permissible under local policy. A nonviable fetus was delivered, following which family consented to a full postmortem examination and case report publication.
Postmortem Findings
The postmortem examination identified a nonviable fetus with multiple congenital abnormalities. External abnormalities included low-set ears, a defect of the left chest wall with protruding lung, liver, bowel loops, and spleen, severe distension of the lower abdomen and initial portion of the umbilical cord (See Figure 6). In addition, there was a thinned/membrane-like abdominal wall, and anal atresia (See Figure 7). Internal abnormalities identified bilateral posterior diaphragmatic defects (See Figure 8) with bowel loops in the right chest cavity. The left chest contained portions of spleen, liver and bowel loops with secondarily displaced, and hypoplastic lungs (lung: body weight ratio = 0.0075, expected ratio = 0.012). Other internal abnormalities included malrotated mesentery and bowels, severely dilated urinary bladder (See Figure 9) with normal draining ureters, and rectal atresia with distended large bowel adjacent. The fetus brain appeared soft and cyanotic. External genitalia comprised a phallic structure (See Figure 7), however internally there appeared to be a uterine structure between the urinary bladder and distended rectum (See Figure 10). Gonadal tissue and internal Mullerian structures were identified showing features consistent with developing fallopian tubes and a uterus.
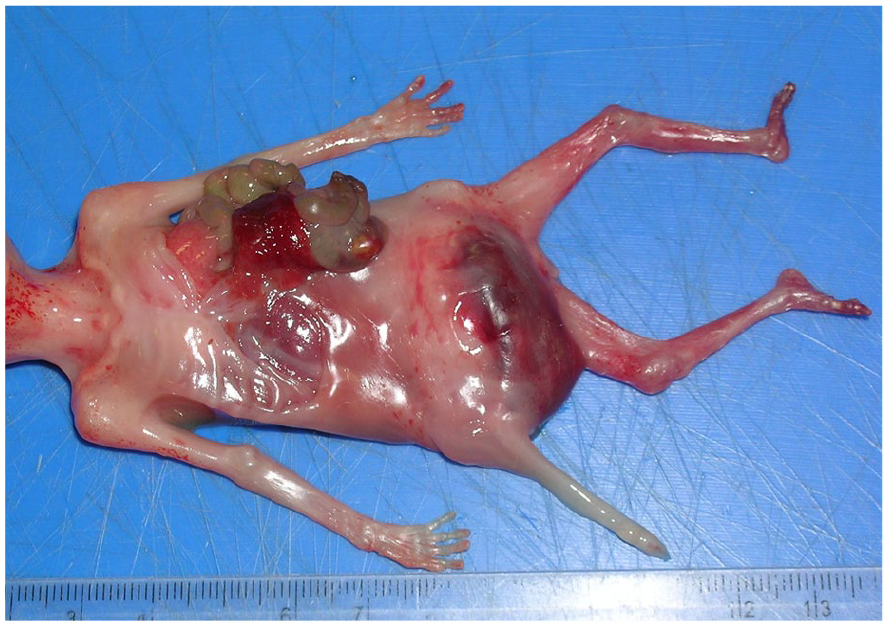
A post-mortem photograph indicated a left chest wall defect with protruding lung, liver, bowel loops, and spleen and severe distension of the lower abdomen and proximal umbilical cord.
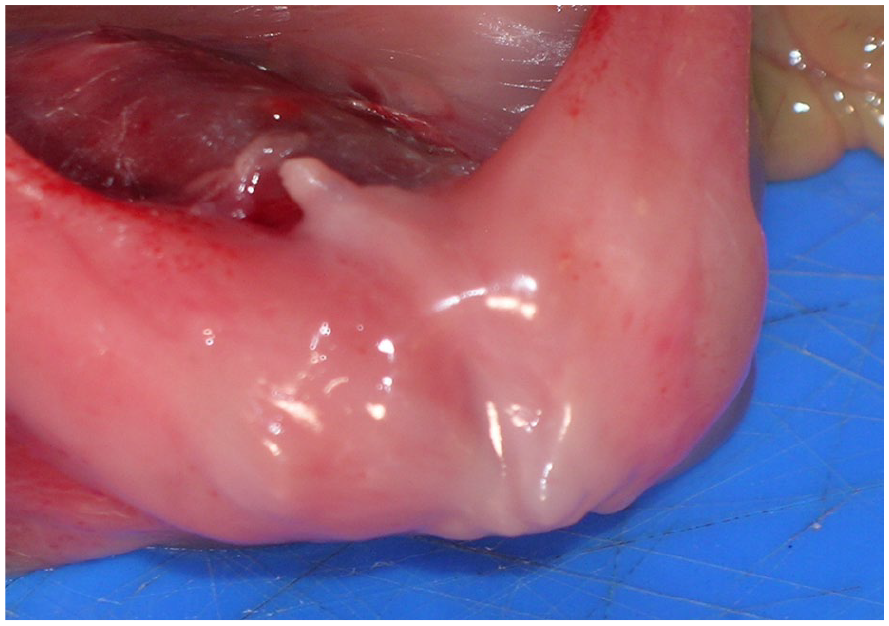
A postmortem photograph of the external genitalia, which comprised a small penis-like structure, absent scrotum, and an imperforate anus.
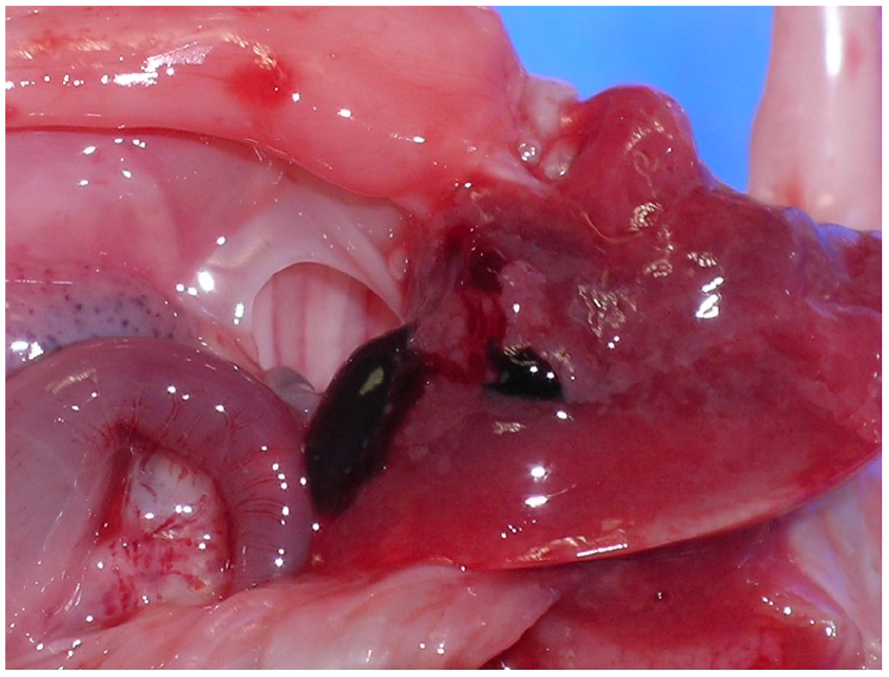
A postmortem photograph documents the posterior defect in left hemidiaphragm.
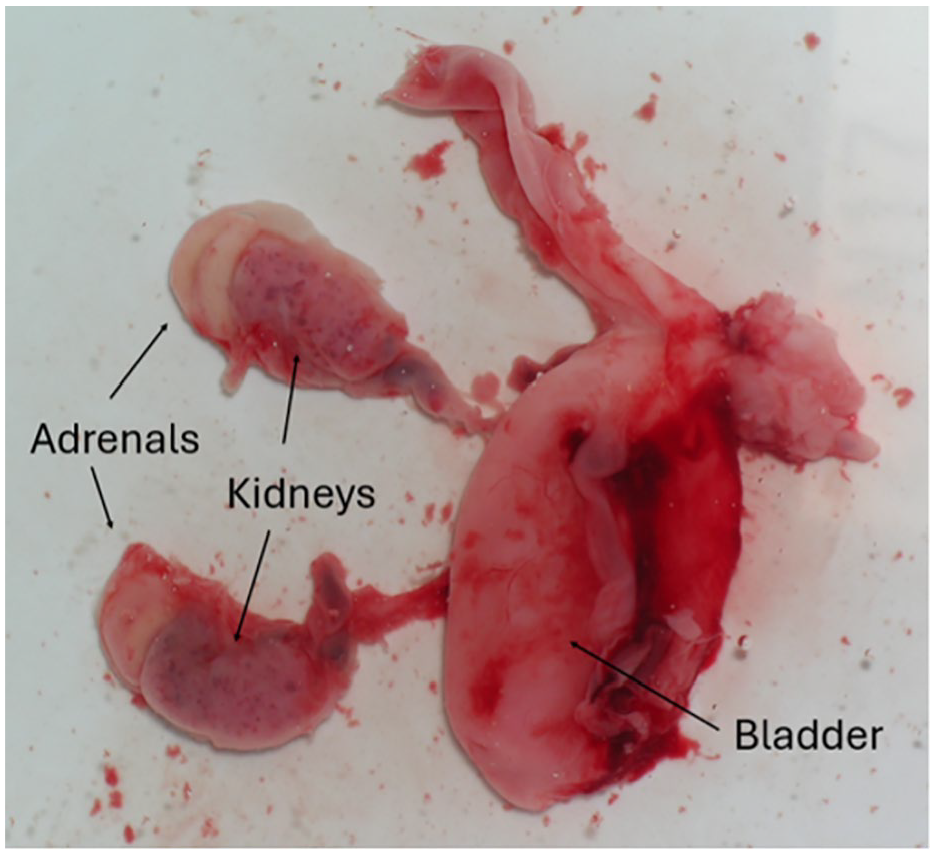
As part of the postmortem investigation, a photograph (taken underwater) displays a distended urinary bladder with attached ureters, kidneys, and adrenals.
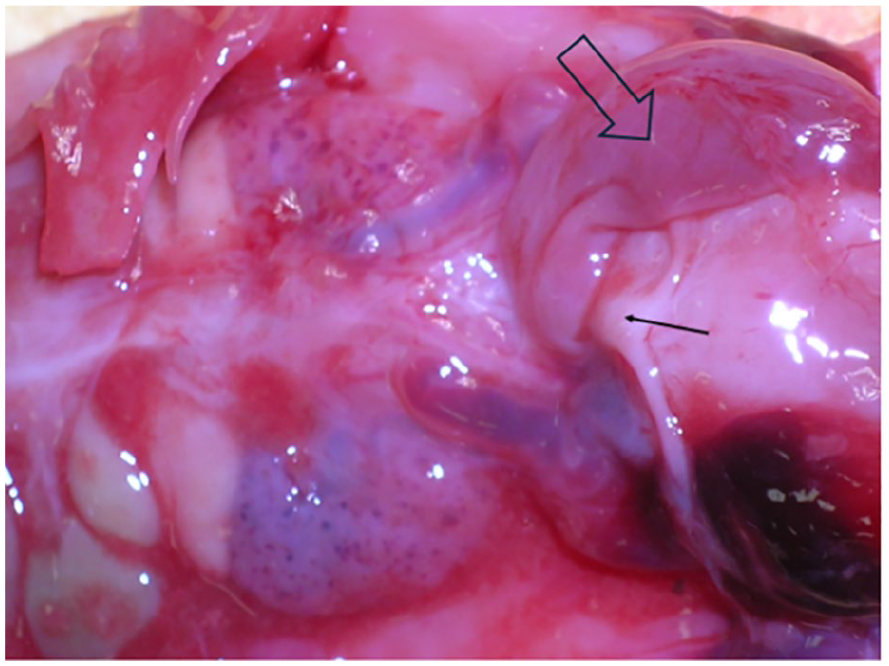
This postmortem photograph documents a Uterine structure (indicated by the black line arrow) located between the urinary bladder and distended rectum (marked by the open arrow).
There were postmortem radiographs taken using a high-fidelity radiographic system with delicate tissue handling and specialized instrumentation. The skeletal survey confirmed bone maturation consistent with a 14-week fetus with 12 right-sided and 13 left-sided ribs. A left lateral chest wall defect containing a soft tissue mass of a serpiginous configuration suggestive of a large bowel was identified, indicating thoracoschisis (See Figure 11). No limb abnormalities were identified although it was noted that these findings are typically described as part of a limb-body wall complex. There were no other fractures or skeletal pathology detected.
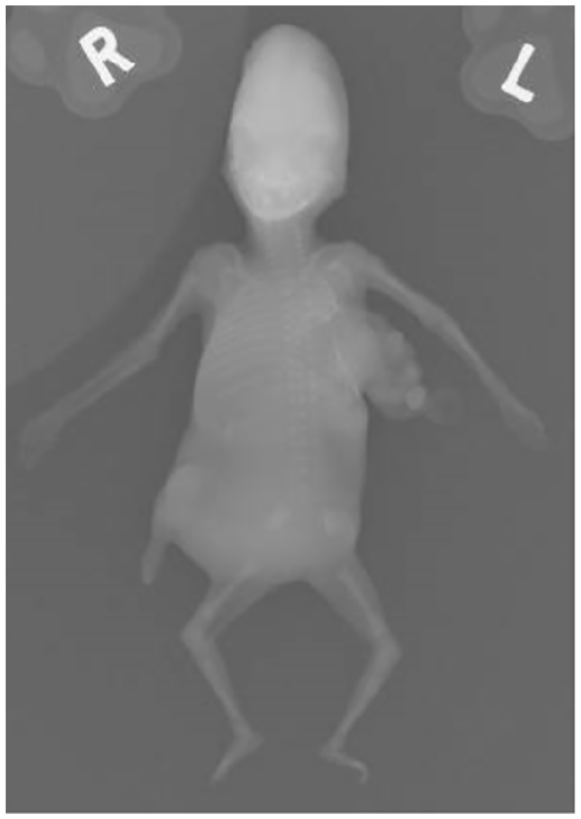
A postmortem radiograph was taken that revealed distorted left sided ribs with thoracoschisis.
The sole abnormality identified in fetal histology was bilateral renal dysplasia. Gonadal tissue showed histology in keeping with ovarian tissue. An external penile urethra, with narrow but patent lumen that extended into the bladder neck and bladder, was identified. More proximal sections showed bladder merging with a structure showing features consistent with a uterus. Placental histology showed patchy villous dysmorphic features along with areas consistent with second trimester pregnancy. Despite the dysmorphic placental features indicating aneuploidy, microsatellite-based testing for chromosomes 13, 18, 21 were normal. Microarray results also failed to detect any known pathological variants but did suggest a 46XX genotype, indicating discordance with the male phenotype identified at postmortem examination. The parents were referred to the clinical genetics department for further counseling. They were offered whole genome sequencing, in response to the fetal finding but it was declined by the family.
Fetal Dysmorphology Multidisciplinary Team Conclusions
The set of multiple anomalies did not fulfill diagnostic criteria of known syndromes, sequences, complexes, or associations. The fetus was identified as female, with internal female genitalia and an external phallic structure which may have been in the spectrum of a cloacal abnormality, DSD, or more extensive diagnosis. The chest wall defect was postulated to be related to delivery, given it was not visible on previous sonographic examinations. It was concluded that this case represented a sporadic of multiple anomalies with empiric recurrence risk of 3% to 5%. The sporadicity of this case was later confirmed when a consecutive pregnancy yielded a fetus with none of these fetal abnormalities.
Discussion
In the case presented, a 14-week fetus was identified as having a sporadic presentation of LUTO, anorectal atresia, exomphalos, CDH, and DSD. Presentation of isolated LUTO around this time is unremarkable; prenatal diagnosis is often made within 20 weeks during routine anomaly sonograms.27,28 Similarly, CDH is often detected during anomaly screening with around two thirds diagnosed by the end of the second trimester. 13 Anorectal atresia is a difficult prenatal diagnosis, with 16% of cases being diagnosed before birth, partly due to sonographic features overlap with other differential diagnoses or may form part of healthy transient structures.29,30 It is therefore difficult to comment on the significance of this presentation of anorectal atresia at this gestational age, although Tapipale et al 31 have previously documented a first trimester imperforate anus. Exomphalos presents after failed re-entrance of bowel back into the midgut, which occurs around the 6-week to 11-week window,32,33 and has previously been reported in the first trimester. 34
It is important to highlight that whilst the fetal dysmorphology team identified CDH as a likely sporadic presentation, it is possible that the diaphragmatic defect may have been secondary to a delivery-related rise in intra-abdominal pressure, as was postulated with the chest wall defect. On previous sonograms, the chest wall was intact, the lungs and heart were normally sited with no mediastinal deviation, and the entire bowel loops were intra-abdominal only, which may suggest a delivery-related etiology. However the postmortem findings remain highly suggestive of CDH and given that detection rate of CDH is around 59% with diagnosis typically made later in gestation, 13 the lack of sonographic features before birth does not dismiss sporadic CDH.
In isolation, the malformations described in this case are not unique. However, there has been no published sporadic case of an early gestation fetus with this combination of fetal malformations. Sporadic urogenital and anorectal malformations in combination with exomphalos have been reported in the complex association OEIS (Omphalocele, Exstrophy of the bladder, Imperforate Anus and Spinal defects). 35 While OEIS shares some features of this case, the lack of spinal defects and presence of a CDH differentiate it into a separate phenotype. The identification of prune belly syndrome (PBS, Eagle-Barrett Syndrome, Triad Syndrome) shares some facets of this case with deficient abdominal wall integrity, urinary tract dilatation, occasional associated cardiopulmonary and gastrointestinal features. 36 In the present case, this fetus lacked the characteristic cryptorchidism of OEIS. The diagnosis of megacystis microcolon intestinal hypoperistalsis syndrome (MMHIS) 37 would fail to encapsulate all the diagnostic features of this case.
Chao et al 38 present a case with some fetal similarities, such as LUTO, alongside an imperforate anus. However, the LUTO cause was megalourethra secondary to penile malformation, as opposed to the urethral narrowing presented in the current case. Erzincan et al 39 provided a case of cloacal dysgenesis causing urogenital and anorectal disturbance with a peculiar ventral disturbance in the form of two-vessel umbilical cord-cyst malformation, without CDH or exomphalos. Krapp et al 40 have documented a first trimester urethral atresia alongside VACTERL association including anorectal atresia, but the fetus presented in this case did not qualify for VACTERL classification.
The microarray findings of 46,XX indicated that the current case also represented a DSD. Prenatal sonographic findings of DSDs, if present, would typically involve atypical or ambiguous genitalia22,23 which were not identified in this case. Prenatal diagnostics of DSDs are possible using invasive genetic testing techniques, such as amniocentesis or chorionic villus sampling, but were not indicated for this pregnancy as there were no identified risk factors. Disorders of sexual differentiation causing male virilization in a 46,XX genotype are largely caused by mutations in enzymes mediating steroid hormone production within the adrenal glands, causing hypersecretion of androgens which lead to virilization in the absence of a Y chromosome.22,23,41 Without whole genome sequencing however, it is not possible to conclude the specific etiology of the DSD present in this case. That the fetus did not show any ambiguous genitalia prenatally and did not age to the point where features of common DSDs, such as Klinefelter or Turner syndrome, could be identified complicates this case further.
The lack of classification into a known syndrome, sequence, complex, or association based on the postmortem conclusions, literature search, and lack of similar case reports, may indicate that this is the first report of such a case. This case report provides initial evidence that the combination of LUTO, CDH, exomphalos, anorectal atresia, and DSD in early pregnancy is possible. Further reports describing similar cases are not currently available but if recorded in future may contribute toward identification of underlying causes or previously unclassified associations between these malformations. However, the lack of reports describing similar cases thus far indicates this is a sporadic entity.
Conclusion
The detection of sporadic LUTO, CDH, exomphalos, anorectal atresia, and DSD, in early pregnancy, is possible and may have been described for the first time, based on available published literature. Analogous patient reports with some degree of similarity have been described but none containing all the malformations identified in this fetus. It is unclear at this stage if associations between these malformations will be identified with further levels of evidence.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Ethical approval for this study was not sought for the present study because all case data were de-identified.
Informed Consent
Informed consent was not sought because all case data were de-identified and/or aggregated and followed the ethics committee or IRB guidelines (Also referred to as the Honest Broker System).