
Abstract
A large experimental campaign has been developed in order to study the passivity of steel embedded in mortar. Mortar samples, containing steel pill (initially polished or precorroded), have been placed in various conditions [relative humidities of 80, 90 and 95%; solution simulating the clay rock underground water of the French candidate site of Bure at two temperatures (25 and 50°C) and in aerated and anoxic conditions]. The passive corrosion behaviour of steel has been assessed after 6 months and after 1 year for each experiment. Gravimetric measurements (average corrosion rate evaluation), X-ray photoelectron spectroscopy and Raman spectroscopy characterisations (identification of the passive film and corrosion product nature) as well as optical and electronic microscopy analyses (corrosion layer thickness and steel/mortar interface elementary analysis) have been performed. Such analyses have been also carried out after 15 days and 1 and 3 months in order to consider the first stages of passivity in mortars. The results show that average corrosion rates determinate for polished steels are typical of passive conditions (<1 μm/year) and vary from 0·4 to 5 μm/year for the precorroded pills. Moreover, iron is detected within the cementitious material, up to 5-50 μm from the metal, indicating the transport of iron species.
Introduction
In the context of French nuclear waste disposal of medium level radioactive wastes, containers and engineered barriers will be built with reinforced concrete. Owing to the alkaline environment, rebars will be submitted to passive corrosion. The present study aimed to determine the kinetics and corrosion patterns that take place during the passive state of steel in cementitious materials in disposal environmental conditions (clay rock environment, water saturated and anoxic).
Experimental
Materials
A first set of metallic samples was embedded in mortar (normalised mortar prepared with a Val d'Azergues CEM I type cement, w/c = 0·5 and s/c = 3):
steel pills (diameter, 16 mm; height, 5 mm): polished or chemically precorroded (pills immersed in H2SO4–Na2SO4 solution during 24 h)
old corrosion product layers (without metallic matrix) coming from ‘Palais des Papes’ in Avignon (France), which have been previously well characterised.1
Mortar samples containing steel pills initially polished or precorroded were placed in 18 different conditions [relative humidities of 80, 90 and 95%; solution simulating the clay underground water (simulated Bure site water, see Table 1) at two temperatures (25 and 50°C) and in aerated and anoxic conditions]. Mortar samples containing old corrosion product layers were stored at 90% relative humidity.
Calculated chemical composition of Bure site water used for experiments on steels embedded in mortar2

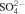
A second set of steel samples (polished or chemically precorroded) has been immersed in solutions representative of three concrete chemical degradation levels (pH 12·6/11 and 9·9) in anaerobic conditions (25°C) (see Table 2).
Chemical compositions of three solutions used for experiments on steels immersed in solution
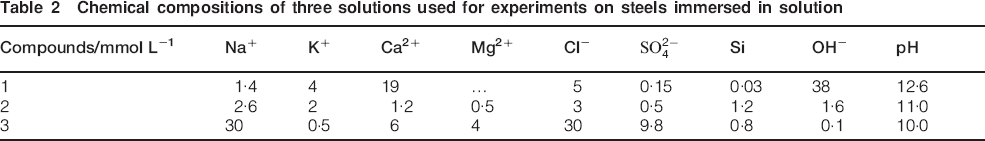
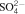
Characterisations were performed after 1, 6, 12 and 24 month tests for pills embedded in mortar and after 1, 2, 4 and 12 month tests for pills immersed in solutions.
Samples dedicated to surface analysis (Raman spectroscopy and SEM-EDS) were cut in order to observe the steel/cement paste interface. They were then mounted in epoxy resin at room temperature. The cross-section surface was prepared by polishing with ethanol, first with SiC (grade 80-4000) and second using diamond paste (3 μm). Observations were therefore representative of only one slice (two sides) of each sample.
X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectroscopy allows the examination of the oxidation states of iron in the passive film. Spectra (XPS) were recorded with a VG ESCALAB 220i XL spectrometer (VG Scientific Society). The X-ray source was the non-monochromatic ray Kα 1,2 of aluminium (1486·6 eV). The beam diameter was ∼6×7 mm. The pass energy was 20 eV. The spectrometer was calibrated in energy to the silver Fermi level (0 eV) and to the 3d 5/2 electronic level of silver (368·3 eV). The low pressure in the analysis chamber (10−10 mbar) allowed to avoid collisions between photoelectrons and residual gas atoms and in situ contamination of the sample surface.
Micro-Raman spectroscopy
In order to identify the nature of the corrosion products present at the precorroded steel/mortar interface, analyses by micro-Raman spectroscopy were performed on the polished cross-sections at Laboratoire de Dynamique Interaction et Réactivité (UMR 7075). A Jobin Yvon-Horiba LabRam Infinity spectrometer equipped with a frequency doubled Nd∶YAG laser at 532 nm was used. The spectral resolution of this set-up is ∼2 cm−1. Micromeasurements were achieved with a ×100 long focus Leitz objective, which gives a beam waist diameter of ∼1·5 μm. The excitation laser power on the sample is filtered at least below 0·1 mW in order to avoid the thermal effect for sensitive iron oxides and oxyhydroxides.3 The phases are identified by comparison with powder standards given by earlier publications.4
Scanning electron microscopy and energy dispersive spectroscopy
Samples were also observed with two different scanning electron microscopes. The first one is a LEO 120, from Cambridge Instruments, with 15 kV accelerating voltage driven by the SAMx MaxView version 4.1.3 software. Energy dispersive X-ray spectroscopy analyses (EDS Delta+, Kevex data acquisition system) were performed using the SAMx IDFix software. The second one is a field emission gun equipped with a hot cathode (MEB-FEG 7000F, JEOL). The acquisition system is a Bruker SDD detector (silicon dry diode) with energy resolution of 127 eV driven by the Spirit software from PGT.
This experimental set-up permitted to analyse the chemical composition of the corrosion products and of the binder. The Si(Li) detectors used for these analyses were equipped with a thin beryllium window in order to detect and quantify oxygen with good accuracy (∼2 mass-% relative error on iron oxides standards). The lower limit of detection of this method is 0·5 mass-% for elements heavier than aluminium and several per cents for oxygen.
Gravimetric measurements
Corrosion products were removed from the steel surface by chemical attack.5 The difference between the initial mass (weight before sample manufacture) and the final one (weight after chemical treatment) gives directly the steel mass loss. This mass loss corresponds to the average corrosion rate for the duration of the test, considering a homogeneous corrosion pattern.
Results
Average corrosion rates and corrosion mechanism
The average corrosion rates were estimated (Fig. 1) from gravimetric measurements performed on polished pills immersed in solutions presenting three different pH values (Fig. 1a) and for polished pills embedded in mortar placed in different environmental conditions (Fig. 1b).
Average corrosion rates deduced from gravimetric measurements for a polished pills immersed in solutions after 1, 2, 4 and 12 months (two values per pH per test duration) and b polished pills embedded in mortar after 12 months in different environmental conditions
Results point out that the corrosion rate is increasing with the decrease in pH values from 12·6 to 9·9. Moreover, the corrosion rate is decreasing with the exposure duration (from 5 to <1 μm/year). This result has been already described by previous authors.6 At this stage, it is difficult to extrapolate these three stages, and the present study proposed a law that describes the evolution of corrosion rate with time.
The corrosion rates determined for pills embedded in mortar placed in different environmental conditions after 12 month exposure are lower than 0·16 μm/year. This emphasises the passive corrosion state of steel that has been already estimated as 0·05 μm/year by Grauer et al.7 Moreover, one has to notice that the passive corrosion rates do not seem to be modified by the presence of 41 mmol L−1 chloride in Bure's water.
The comparison between corrosion rates determined for pills immersed in solution pH 12·6 and for pills embedded in mortar in deaerated conditions (25°C) after 12 months points out that the corrosion rates are similar in solution and in mortar for polished steels (∼0·1 μm/year). This result indicates that the system is kinetically limited by electrochemical processes within the passive film.
Nature of passive film
Passive film analysis performed by XPS for polished pills embedded in mortar after 1 year (Fig. 2) highlighted several points:
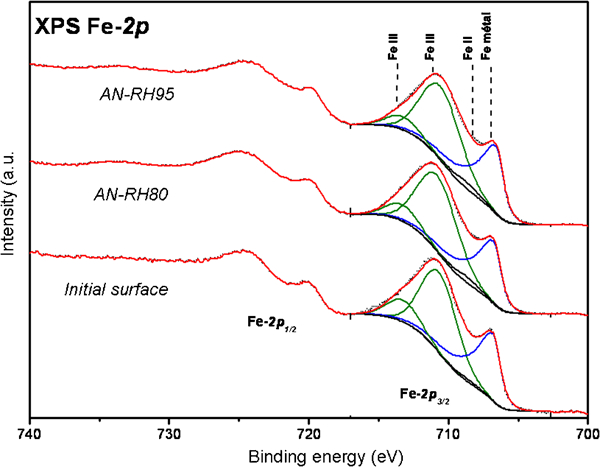
Superposition of XPS Fe 2p spectra for initial surface (before being embedded in mortar) and for surfaces embedded in mortar after 12 months at 80 and 95% relative humidities in anoxic conditions
the iron metallic peak is present for all conditions; this indicates that the passive layer thickness is lower than 10 nm
a deconvolution of the iron oxide signal allows to detect mainly iron III within the passive film; this result had been pointed out previously for steels immersed in alkaline solutions8
all XPS spectra were similar to the initial ones obtained before embedding the steel (after passivation in atmosphere); this means that the passive film probably does not evolve during the exposure in mortar.
Nature of corrosion products
As far as precorroded pills are concerned, Raman analysis performed after the precorrosion treatment highlighted the presence of goethite (α-FeOOH), ferrihydrite (5Fe2O3.9H2O) and lepidocrocite (γ-FeOOH) within the corrosion product layer.
Steels embedded in mortar placed in different relative humidities (80, 90 and 95%) in aerated conditions present the same corrosion product layers as initially, with mainly goethite and lepidocrocite phases (Fig. 3 and Table 3).
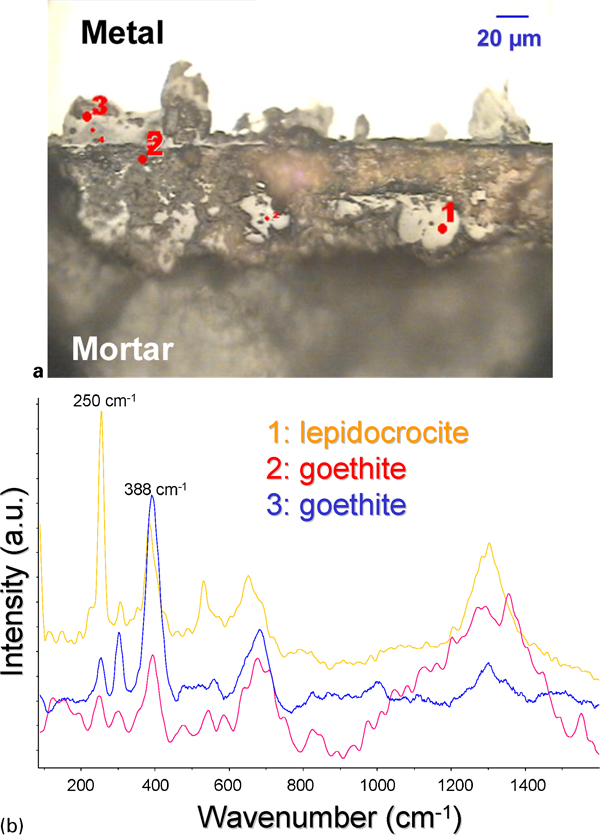
Sample embedded in mortar exposed during 12 months at 80% relative humidity in aerated conditions
Iron oxide phases detected within corrosion product layers formed on steel pills embedded in mortar placed in different conditions in relative humidity and in simulated Bure site water

The corrosion product layers detected for steels embedded in mortar exposed at RH in anoxic conditions and in simulated Bure site water are quite different from the initial ones. In particular, magnetite (Fe3O4) and haematite (α-Fe2O3) species seem to appear within these layers after 2 years of test (Fig. 4 and Table 3).
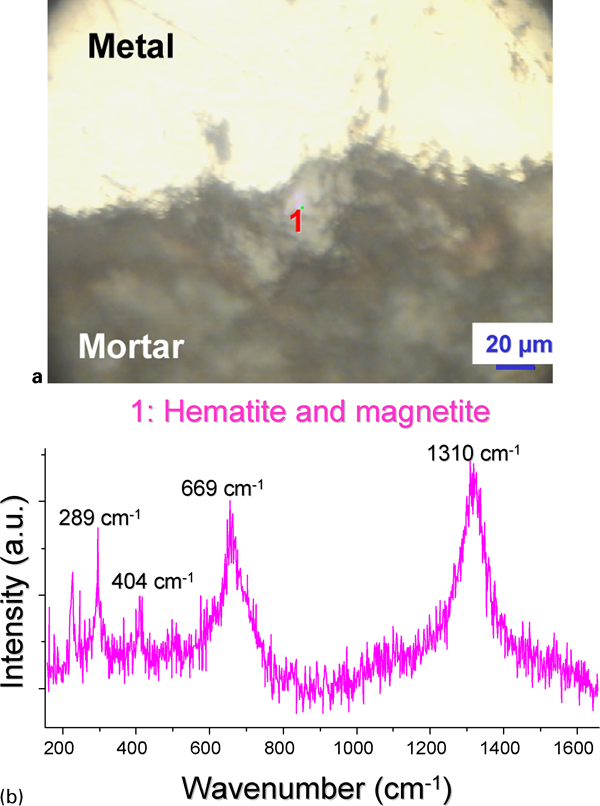
Sample embedded in mortar exposed during 2 years at 95% relative humidity in anoxic conditions
The appearance of iron oxides such as magnetite and haematite will have to be further investigated and explained. One explanation could be that as the mortars are manufactured in aerated conditions and placed in anoxic chambers after 24 h, the oxygen entrapped within the material could ensure the stability of initial corrosion products during a certain time. When the anoxic conditions are reached in the whole mortar sample, more reductive oxides are able to form due to thermodynamic considerations.9
Steel/mortar interface
Analyses by SEM-EDS of the steel/mortar interface highlighted two kinds of corrosion patterns.
The first typical corrosion pattern is observed on precorroded samples. In this case, the metal is surrounded by a double corrosion layer (metal–‘layer 1’–‘layer 2’–mortar) before the mortar matrix (Fig. 5). ‘Layer 1’ is always around 20-40 μm thick and contains 30-50 mass-%Fe and 5-20 mass-%Ca. ‘Layer 2’ is 10 μm thick and contains 5-20 mass-%Fe and 30-50 mass-%Ca.

Micrographs of selected zone for SEM-EDS analysis on precorroded pill embedded in mortar immersed in simulated Bure site water (25°C) in anoxic conditions during 12 months
The second configuration that is detected is found on precorroded samples (zones for which there is no corrosion product layer) as well as on polished samples (Fig. 6a). In this case, the metal is surrounded by ‘layer 2’ (10 μm thick and contains 5-20 mass-%Fe and 30-50 mass-%Ca) at the mortar interface. This configuration was also detected at very short term (1 month) on polished and precorroded samples as well as in ‘old corrosion product layer’/mortar interface (Fig. 6b).

Micrographs and Fe contents (SEM-EDS) of selected zone on a polished pills embedded in mortar immersed in simulated Bure site water (50°C) in aerated conditions during 12 months and b old corrosion product layer embedded in mortar exposed at 90% relative humidity during 1 month (aerated conditions)
These results indicate that the corrosion pattern is stable between 1 and 24 months. It does not evolve with time or with environmental conditions.
Conclusions
The large experimental campaign developed in order to assess the passivity of steel embedded in mortar allowed to draw the following conclusions.
The estimated average corrosion rates are typical of the passive corrosion state. The passivity of steels embedded in concrete is verified for all conditions tested. Moreover, the electrochemical processes within the passive film are the main processes to be taken into account in the modelling.10,11
Iron and calcium transport processes take place at the metallic sample/mortar interface. For instance, in the case of precorroded steel, the initial corrosion product layer is ‘polluted’ by 5-20 mass-%Ca. On the other side, in the case of polished steel and also of old corrosion product layer embedded in mortar, the mortar surrounding the metallic part is ‘polluted’ by 5-20 mass-%Fe. These transport phenomena will have to be further explained as well as the nature of the phases containing Fe and Ca.
The nature of corrosion products present at the precorroded steel/mortar sample evolves in specific conditions (deaerated and water saturated environments) until phases typical for reductive conditions (magnetite and haematite). Further studies will have to be carried out to explain this modification.
Up to now, a part of the samples is still under corrosion process in the various selected conditions. Characterisation of such iron/mortar system after 36 and 48 months are already planned.
Footnotes
Acknowledgements
The authors acknowledge the financial support provided by CEA, ANDRA and EdF (French ‘GL ESC’).