
Abstract
Magnesium alloys are promising orthopaedic bioresorbable implant candidates, however, their inherent rapid corrosion rate in physiological media currently limits their clinical applications. In this work, the in vitro corrosion of a series of Mg–xCa and Mg–3Zn–yCa alloys (wt-%) was systematically studied. These compositions were selected so the alloys could be comprised of biocompatible elements and to explore the role of Ca in the alloy itself upon subsequent calcium phosphate (CaP) coating efficiency. A simulated body environment was reproduced via minimum essential medium (MEM) and exposure in a CO2 incubator at 37°C. The effect of Ca and Zn additions on the corrosion rate of Mg was examined, indicating the corrosion rate increases with Ca additions, while adding Zn to Mg–Ca alloys decreases the rate of corrosion. The impact of a biomimetic CaP conversion coating applied onto these alloys indicated that although the coating alters the corrosion rates, the effect is dependent on the substrate alloy composition.
Introduction
When considered for orthopaedic applications, magnesium (Mg) and its alloys have several unique advantages over conventional biomaterials such as titanium alloys, stainless steel and cobalt–chromium alloys. The elastic modulus of most Mg alloys (40-45 GPa)1 is closer to that of human bone (40-57 GPa), which can significantly reduce problems associated with the stress shielding effect,2 while the density of Mg and its alloys (∼1·7 g cm−3) is close to that of human calvarium bone (1·75 g m−3).3 In addition to Mg, which is one of the major elements found in bone tissue,4 perhaps most crucially, Mg alloys also present the unique opportunity to provide an orthopaedic implant material that will safely biodegrade inside the body.5
It is posited that biodegradation products of Mg based biomaterials are likely to be advantageous rather than harmful, and this is a reasonable design criterion. Mg itself is an essential element to the human body,6,7 being biologically active and with a beneficial effect on cell growth and mineralisation of bony tissue through its control of hydroxyapatite formation.8 In addition, alloying elements, such as Ca9 and Zn,10 have been shown to be beneficial in osteoblast differentiation and bone precursor formation. Consequently, in this study, we investigate alloys containing such biocompatible elements, namely, Zn11,12 and Ca,13 – 15 which were combined with Mg to form binary/ternary alloys. The benefits of Zn and Ca include: corrosion products that are non-toxic for the surrounding tissue;5,16,17 as a major component in the bone matrix, Ca may accelerate bone growth, reducing healing time;9 and Zn and Ca are essential nutrient elements and are efficiently utilised by the body (15 and 800 mg/day respectively).7,10 Finally, both Ca and Zn have been shown to significantly improve the mechanical properties of Mg, allowing for a wider range of orthopaedic applications.18
Many Mg alloys are noted to rapidly corrode at the physiological pH (7·4-7·6) or when chloride (Cl−) ions are present. Consequently, the possibility exists that an implant can dissolve and loose its mechanical integrity before it has carried out its intended function, not providing sufficient time for fractured bone to heal.2,4 In addition, hydrogen gas formation, produced by the corrosion process of Mg (and in particular when corrosion is vigorous), may aggregate in the vicinity of implants, damaging the integrity of the fracture sites.19,20 Further alloying is a method that may reduce the corrosion rate of Mg, however, the addition of potentially toxic elements such as aluminium or rare earth metals may dramatically effect the biocompatibility of Mg alloys and present short and long term concerns for the health of the patient.12
Based on recent works, the expected corrosion of Mg–Ca–Zn alloys may be too rapid for some biomedical applications, where the implants are required to maintain their mechanical strength and integrity over a time scale of 12-18 weeks until the bone tissue heals and is replaced by natural tissue.21 Hence, this study is a discrete effort in investigating the ability to slow the corrosion of Mg–Ca–Zn alloys. Calcium phosphate (CaP) coatings are investigated, as Ca and phosphorus (P) are the primary components of bone minerals. A CaP coating that includes a crystalline hydroxyapatite film, which is the most stable form of CaP compounds, may be able to not only offer corrosion protection to the subsurface but also improve bone cell growth and speed the formation of new bone.22 – 26 Herein, we aim to qualify and quantify the biocorrosion of a series of Mg–xCa and Mg–3Zn–yCa (x = 1·34 and 5 wt-%; y = 1·34, 5, 10 and 16·2 wt-%) alloys, under physiological conditions, with and without a CaP coating applied by a two-step conversion coating process.27 This will also allow the ability to comment on the role of substrate Ca content on the subsequent coating efficacy.
Experimental
Materials preparation
The alloys studied herein were custom produced via induction melting and casting in an inert atmosphere. To study the corrosion response as a function of variations in Ca concentration, the alloys tested include a binary Mg–Ca series and a ternary Mg–Zn–Ca series with constant Zn levels (to again study the role of varying Ca). Alloys were produced by blending of high purity Mg (99·99wt-%) and an Mg–28Ca wt-% master alloy (Timminco, Toronto, Canada). Melting was performed under pure argon (0 grade) using an induction furnace, and the alloys were cast into a graphite coated cylindrical mild steel crucible to produce 60 mm diameter, 60-70 mm tall ingots of ∼250 g. The melt temperature was held at 750°C for 15 min, during which the melt was stirred for 1 min every 2 min. The melt was then cooled at an average rate of 1°C s−1.
Microstructural characterisation
Samples were ground with 1200 grit silicon carbide (SiC) paper, then polished with 3 and 1 μm diamond slurries, followed by a final soft pad and slurry polish (BuehlerVR ChemoMet and BeuhlerVR Master-Met) to obtain a finish of 0·05 μm. The as polished surfaces were observed to display different phases and grain boundaries without further etching. The surfaces were analysed using an FEI Phenom scanning electron microscope (SEM). The actual alloy chemical composition was determined with an inductively coupled plasma optical emission spectrometer (Spectrometer Services, Coburg, VIC., Australia) and reported in Table 1.
Inductively coupled plasma optical emission spectrometer analysis of Mg alloys used in this work

Energy dispersive X-ray (EDX) analysis of CaP coated Mg alloys

Conversion coating process
Mg alloy specimens (1×1×1 cm3) were progressively ground to 1200 grit and sonicated in acetone and ethanol before any experiment. The conversion coating process required two steps and is of the form outlined by Chen and co-workers27 for pure Mg. The primary coating solutions contained 0·017M calcium nitrite [Ca(NO3)2] and 0·01M sodium phosphate (Na3PO4) and were adjusted with nitric acid (HNO3) to pH 3·0. The coating process was conducted by immersing Mg alloy specimens into the heated coating solution at 65°C for 2 min. Following this, a secondary coating/post-treatment was carried out in a 10 g L−1 NaOH solution at 80°C for 4 h. All chemicals used in this work were analytical grade (Sigma-Aldrich).
Surface analysis
The surface morphology of the coatings was observed using SEM equipped with EDX spectroscopy (Philip XL30). Surface phase compositions were examined by an X-ray photoelectron spectroscope (XPS, K-Alpha, Thermo VG Scientific, UK). The photoelectrons generated by Al Kα (1486·6 eV) primary radiation (20 kV, 15 mA) were analysed with a hemispherical analyser, and the core level XPS spectra for Ca 2p, Mg 2p, Zn 2p, P 2p, O 1s and C 1s were measured. The measured binding energy values were calibrated by the C 1s (hydrocarbon C–C, C–H) of 285 eV.
In vitro biodegradation/corrosion
To reproduce the human body conditions, a CO2 incubator (Sanyo) and a simulated body fluid, minimum essential medium (MEM, Sigma-Aldrich M0643), were used.15 The MEM contained inorganic ions in concentrations very close to that of human blood plasma. The sterile Sanyo incubator maintained the MEM at the human body temperature of 37±0·1°C and provided a constant CO2 atmosphere of 5%, which maintained the pH of the media at 7·4±0·05. For mass loss immersion tests, the exposed surface area was 1 cm2. The samples were incubated up to 24 h, after which a 7 wt-% dilute HNO3 solution was used to remove the corrosion products.
The hydrogen volume evolved during exposure was also collected using a funnel placed over the specimen to ensure the collection of hydrogen gas from the specimen surface. A burette with a total volume of 23 mL was mounted over the funnel and filled with MEM. The connections of funnel–burette and burette–peleus ball were sealed using Parafilm tape to avoid leakages.
Electrochemical tests employed the use of a flat cell (PAR) that contained ∼300 mL of MEM to which 1 cm2 of sample (working electrode) was exposed. The reference electrode was a saturated calomel electrode, and the counter electrode was titanium mesh. Open circuit and potentiodynamic polarisation tests were executed using a Biologic VMP-3Z potentiostat and a potentiodynamic polarisation at a sweep rate of 1 mV s−1. Specimens were allowed to stabilise for 10 min to reach a suitably stable open circuit potential before commencing polarisation. The i corr values were determined using a Tafel type extrapolation of the polarisation curve using EC-Lab software V10·0 (Biologic).
Results and discussion
Effect of alloying elements on corrosion of Mg alloys
Microstructure of Mg–xCa and Mg–3Zn–yCa alloys
The chemical compositions of the alloys were characterised via inductively coupled plasma optical emission spectrometer (ICP-OES) analysis to determine the actual amount of each alloying element. The results are summarised in Table 1. Generally, the nominal amount of alloying addition represents an upper alloying limit, with the analysed mass percentages of Zn and Ca found to be in agreement with the expected amount of each alloying element.
The microstructure of the as polished Mg–xCa and Mg–3Zn–yCa alloys (Fig. 1) demonstrates that for all alloys, a second phase was formed, with the exception of Mg–3Zn. Mg–xCa binary alloys typically consist of a dendritic primary α-Mg matrix and intermetallic eutectoid made up of α-Mg and Mg2Ca.28,29 Increasing the Ca content of the Mg–xCa binary alloys induces refinement of both α-Mg and Mg2Ca particles.28 For the Mg–3Zn binary alloy, some Mg–Zn intermetallic phases exist in the primary α-Mg matrix due to the high solid solubility of Zn (6·2 wt-%) in Mg alloy.30 With increasing Ca additions to the Mg–Zn system, both the volume fraction and the particle size of the Mg–Zn phase declines, while the Mg2Ca content and particle size increase.28

Images (SEM) of microstructure of various Mg–xCa and Mg–3Zn–yCa alloys (wt-%)
Corrosion of uncoated Mg–xCa and Mg–3Zn–yCa alloys in MEM
As a biodegradable implant, it is crucial to understand and control the corrosion behaviour of Mg alloys under physiological conditions. However, as long as a suitably slow degradation rate is achieved, the corrosion products of Mg and the above mentioned alloys would not be toxic to animal body.11
As shown in the surface morphology of the uncoated Mg alloys after 24 h exposure to MEM (Fig. 2a), all surfaces were covered with corrosion product. In the case of Ca rich alloys (wt-%⩾5), a large amount of corrosion product is observed,28 which is a consequence of a sustained high corrosion rate. For Mg–16·2Ca, the entire sample was converted to corrosion product. The mass loss rate (mg cm−2/day) of all the alloys after 24 h exposure to MEM (Fig. 3) indicates that a more rapid corrosion rate occurred for the Mg–5Ca, Mg–10Ca and Mg–16·2Ca specimens in comparison to all the other alloys, which stands in agreement with the observation in Fig. 2.

Photographs of a bare Mg–3Zn–yCa and Mg–xCa alloys and b CaP conversion coated Mg–3Zn–yCa and Mg–xCa alloys all before and after exposure to MEM for 10 h
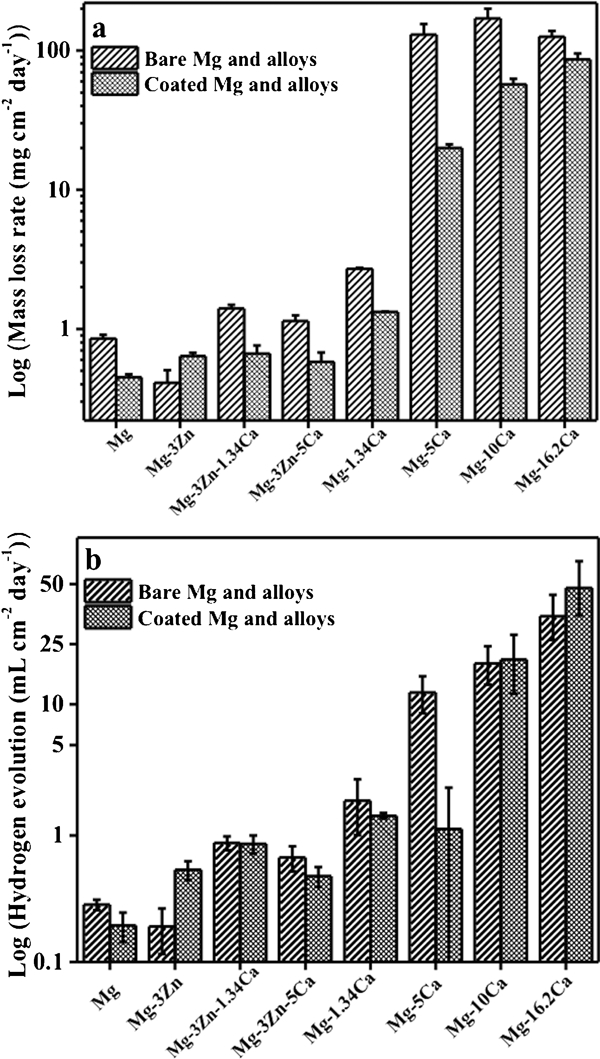
Measured a mass loss rate and b hydrogen evolution rate of Mg alloys after exposure to MEM for 24 h
For the binary Mg–Ca alloys, the corrosion rate is found to decrease initially when the Ca content rises from 0 (pure Mg) to 1·34 wt-%, thereafter increasing rapidly with Ca content until 10 wt-% and finally decreases slightly as the Ca concentration approaches 16·2 wt-%. The greatest mass loss rate of 170 mg cm−2/day was experienced by the Mg–10Ca alloys (Fig. 3a, the differences are statistically significant). The values from all Mg–xCa alloys, except for Mg–1·34Ca, exceed 100 mg cm−2/day, which is commonly assumed too rapid to consider any practical biomedical applications.15,28 Mg–1·34Ca displays a relatively low corrosion rate compared to the other Mg–Ca binary alloys, with a mass loss of 2·72 mg cm−2/day. Analysis of the hydrogen evolution of the alloys after 24 h exposure to MEM (Fig. 3b, the differences among Ca rich alloys are not statistically significant) reveals a strong correlation with the rates as found via mass loss measurement. The Mg2Ca second phase has previously been shown to be more electrochemically active than the primary α-Mg,28 thus dissolving at higher rates (Fig. 4). The influence of the amount of Ca in Mg–xCa alloys is also illustrated by polarisation curves (Fig. 5a), where it can be seen that the cathodic reaction rates increase with increasing Ca content (curves marked as 1, 3, 5 and 7) due to a higher presence of the secondary phase. The anodic reaction of Mg–1·34Ca demonstrates a breakdown potential (marked with an arrow). This is the potential at the end of the passivation region, the point at which the metal suffers pitting corrosion.31 For the Ca rich alloys, no apparent passivation region could be observed, indicating that they are more susceptible to corrosion due to the existence of a higher content of the secondary phase efficient cathode.13 For comparison, the corrosion parameters E corr and i corr are calculated and presented in Fig. 5c. It can be seen that E corr shifted towards lower potentials with increasing Ca additions, indicating the Ca additions enhance the anodic dissolution. This is in agreement with the trend in i corr, where the greater the Ca addition, the higher the corrosion rate.

Microstructures of uncoated Mg–1·34Ca before and after exposed to MEM for 24 h
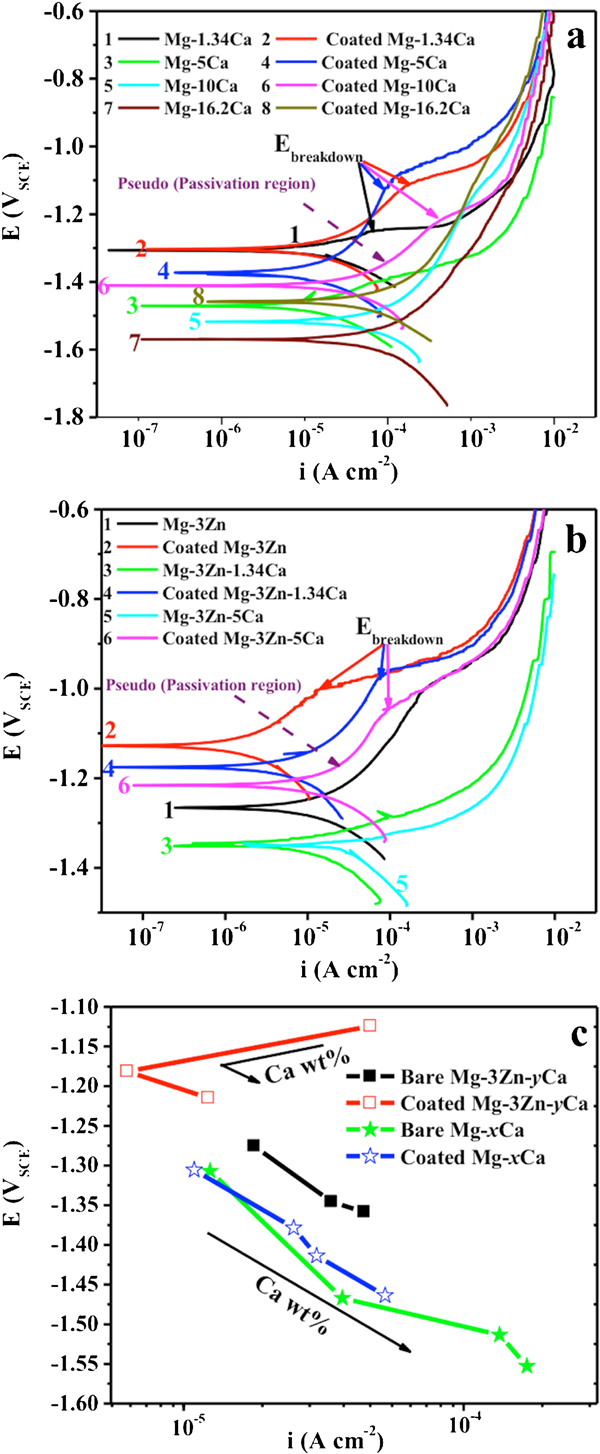
a, b potentiodynamic polarisation curves of the bare and coated Mg–xCa and Mg–3Zn–yCa alloys in MEM and c diagram of corrosion potential E of the bare and coated Mg alloys in dependence of corrosion current i in MEM, calculated by Tafel extrapolation
For the Mg–3Zn–yCa ternary alloys, the mass loss rate increased with a rising Ca content (Fig. 3a). Conversely, Zn addition decreased the corrosion rate by strengthening the rate of anodic dissolution.11,32 Adding 3 wt-% Zn into pure Mg results in a corrosion rate of 0·41 mg cm−2/day, less than half that of pure Mg (0·85 mg cm−2/day). Compared to Mg–5Ca alloy, Mg–3Zn–5Ca alloy displayed a mass loss rate of 100 times lower. This emphasises the performances of Zn in decreasing the corrosion rate. The polarisation curves of the uncoated Mg alloys and the influence of the alloying element Zn on their corrosion behaviours (Fig. 5) emphasise that the cathodic and anodic branches display comparable slopes and following similar reaction rates. A pseudo-passive region is observed for the anodic branch of the polarisation curve of Mg–3Zn only. The Ca alloying element in the Mg–3Zn system is found to shift E corr towards less noble values (refer to curves 1, 3 and 5 in Fig. 5b and c). Additionally, the cathodic and anodic reactions are increased by alloying Mg–Zn with Ca primarily due to the presence of the Mg2Ca secondary phase and the higher surface area ratio of this phase to the Mg matrix that makes it susceptible to microgalvanic corrosion. The calculated corrosion current densities of Mg–3Zn–yCa alloys are in the range of 10−5–10−4 A cm−2. The lowest i corr was found for the Mg–3Zn alloy (1·79×10−5 A cm−2).
Effect of CaP coating on corrosion rate of Mg alloys
Photographs of the coated specimens before and after being exposed to MEM are shown in Fig. 2b, with the coated samples displayed in matte colours. Several small bright dots are visible, which homogeneously spread all over the surface. These dots are localised CaP precipitations. After MEM exposure, localised corrosion took place during the hydrogen evolution that can be seen by shining spots on the surfaces. Local breakdown (pitting corrosion) may occur due to the rough coating's low packing density. Meanwhile, the volume of corrosion product in the case of Mg–10Ca and Mg–16·2Ca is very large, as in the case of the uncoated Ca rich alloys (Fig. 2).
The CaP film is ∼5 μm thick (Fig. 6a). The surface morphologies of the coated samples (Fig. 6b and d) are found to be similar: the specimen are covered with a thin, loose and rough layer consisting of particles that are much smaller than the grain size of the alloys (∼40 μm). XPS analysis of the coatings reveals that the binding energy values of Ca 2p, P 2p and O 1s are 347·5, 133·1 and 531·6 eV respectively. Magnesium hydroxide [Mg(OH)2] was also formed by the Mg 2p signal at ∼49·9 eV. Consequently, it can be concluded that the final coating is a mixture of Ca–P and Mg(OH)2. The binding energy values of Ca and P are not significantly affected by the difference in chemical composition of the alloys. However, EDX analysis indicates the presence of P, the primary indicator for the CaP coatings, and its content be closely related to the alloy composition (Table 2). The maximum amount of 1·33 wt-% P was detected for Mg–10Ca and Mg–16·2Ca, and the minimum of 0·21 wt-% for Mg–3Zn. This is attributed to an increased number of Ca ions in the vicinity of these Ca rich alloy surfaces, allowing more CaP to be deposited. Since the equilibrium of dissolution–precipitation of CaP was broken, it is prone to having more Ca2+ and 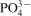 ions deposited onto the surface.
ions deposited onto the surface.

a images (SEM) of cross-section of coated Mg–1·34Ca, b surface morphology of coated Mg–1·34Ca, c surface morphology of coated Mg–1·34Ca after exposed to MEM for 24 h, d surface morphology of coated Mg–3Zn–5Ca and e surface morphology of coated Mg–3Zn–5Ca after exposed to MEM for 24 h
Polarisation curves (Fig. 5a and b) are characterised by a passivation region and breakdown potential for coated samples. Compared to the specimens without coating, both E corr and i corr of the coated samples are more ennoble (Fig. 6c), revealing that the coating efficiently protected all the alloys against corrosion attack, with the exception of the Mg–3Zn alloy. The protection efficacy of the coating can be seen in Fig. 7. The coating worked best on Mg–5Ca while reducing the corrosion rate of Mg–16·2Ca the least. However, for Mg–3Zn alloy, the coating process provided no positive effect on the measured corrosion rate. This is hypothesised to be due to the lower rate of anodic dissolution of the Mg–3Zn alloy when exposed to acidic coating bath, subsequently impacting on the efficacy and quality of the deposited CaP film.
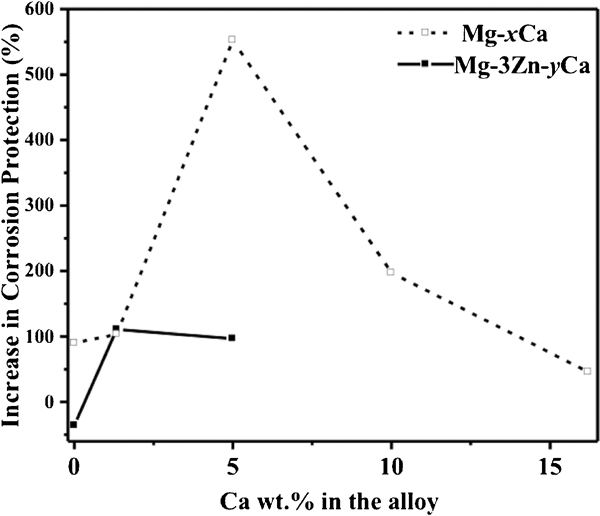
Increase in corrosion protection of Mg and alloys due to CaP coatings. Increase in corrosion protection % = (ML uncoated−ML coated)/ML coated×100% (where ML uncoated is mass loss of uncoated alloys after exposure to MEM, and ML coated is mass loss of coated alloys after exposure to MEM)
General discussion
We have demonstrated that increasing the Ca content beyond the solid solubility limit (1·34 wt-%) significantly increases the corrosion rate of Mg, while adding Zn to Mg moderates the corrosion rate. Although Ca is shown to increase the corrosion rate, it also typically improves the mechanical properties of pure Mg, including creep resistance and ultimate tensile strength.13,33 To comply with human implant requirements, the ideal alloy is a compromise between a suitable corrosion rate and the correct mechanical properties. The addition of Zn will not increase the corrosion rate of the α phase of Mg alloys if Zn forms a solid solution with the α-Mg matrix phase.34 However, a discrete intermetallic precipitate normally results in microgalvanic corrosion and consequently causes an increase in corrosion rate. For the alloys investigated, the galvanic acceleration from Zn rich particles is not possible since Zn is below the solubility limit and does not increase the corrosion rate.
The degradation of coated Mg alloys in MEM can be explained as follows. Firstly, the CaP coating reacts with MEM and partially dissolves.35 Secondly, MEM contacts the Mg substrate through the loose structure of the CaP film, resulting in corrosion and the release of Mg2+ ions; these ions precipitate with Ca and 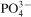 ions to form Mg containing CaP on the substrate.36 Finally, as the Mg substituted CaP is not thermodynamically stable, the locally accumulated Mg2+ further jeopardises the integrity of the CaP coating37 and induces pitting corrosion of the Mg substrate, as revealed in Fig. 6c and e.
ions to form Mg containing CaP on the substrate.36 Finally, as the Mg substituted CaP is not thermodynamically stable, the locally accumulated Mg2+ further jeopardises the integrity of the CaP coating37 and induces pitting corrosion of the Mg substrate, as revealed in Fig. 6c and e.
The effect of the two-step coated CaP films on the corrosion rate of the tested Mg alloys can also be observed in Fig. 8, where, unlike the hydrogen evolution ‘rate’ in Fig. 3b, a cumulative representation of hydrogen evolution versus exposure time to MEM is depicted. This can be divided by the days to give rate, however, more importantly, it demonstrates that at the initial stage, the coating decreases the amount of hydrogen evolved and delays the biodegradation (albeit for a finite period). This is of paramount importance for a new implant following implantation since the development of early cytoxicity from hydrogen gas can be avoided. After ∼7 days, the hydrogen evolution rate seems to track with the uncoated specimen. Such a delayed onset of the degradation process is critical to a biodegradable implant as the implant needs to fully function for a certain period of time before the bone tissue starts healing.38 Obviously, such coatings are imperfect barriers to corrosion, not completely ceasing degradation. However, if they were perfect barriers, they would be unsuitable for biodegradation, so the limited protection may actually be ideal for solving early rapid corrosion than allowing a predictable dissolution to occur.
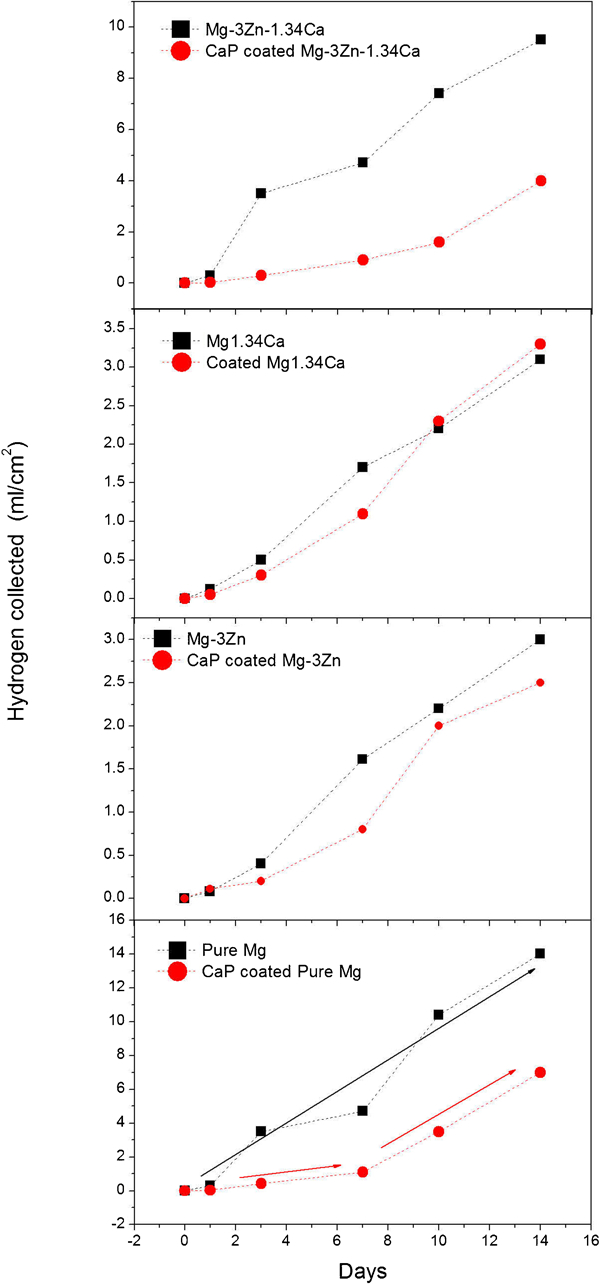
Measured hydrogen evolution volume of uncoated and coated Mg and alloys versus exposed time to MEM
Conclusions
In this work, the in vitro biocorrosion of Mg–xCa and Mg–3Zn–yCa alloys (x = 1·34 and 5 wt-%; y = 1·34, 5, 10 and 16·2 wt-%) was studied. Characterisation of Mg–xCa alloys displays the formation of eutectic α-Mg and Mg2Ca phases. For the Mg–3Zn–yCa ternary alloys, Zn was primarily found to reside within the secondary phase. The corrosion behaviour of the alloys under physiological conditions demonstrates that alloying Mg with increasing levels of Ca increases the corrosion rate when a secondary phase is formed, while Zn lowers the corrosion rate (at the iso-concentration of 3 wt-%) when added to Mg–Ca based alloys.
The impact of a CaP conversion coating was shown to be dependent on the substrate composition. In all cases, the CaP coating was able to reduce the rate of corrosion in Mg–xCa alloys; however the coating efficacy decreased when the Ca content was >5 wt-%. This was most likely due to the difficulty in obtaining a defect free coating for such reactive alloys and the increased content of Ca in the substrate having little impact on promoting an improved CaP coating. The CaP coating was not effective in reducing the corrosion rate of Mg–3Zn, however, for Mg–3Zn–yCa alloys, a reduction in corrosion rate was observed. It was found that the CaP coatings did not serve the role of an exceptional barrier coating, since they did not dramatically reduce the rate of corrosion. However, they did show significant merit in stifling the early stages of corrosion by retarding the amount of hydrogen evolved upon the alloys exposed to MEM. This is likely to be important for real implants, and additionally, the presence of any CaP is also clinically beneficial, though these two points will require future in vivo work to confirm.
Footnotes
Acknowledgements
The following are acknowledged for their financial support: the CAST Co-operative Research Centre, the Australian Research Council (Centre of Excellence for Design in Light Metals), the Go8-DAAD scheme (HK, SV, NB), the MRA award (NB) and the Australian Research Council Post-doctorate Fellowship (DN). The alloys studied were produced at the University of Canterbury (New Zealand) by NTK.