
Abstract
Materials informatics is based on the integration of tools for generating, classifying, analysing and disseminating knowledge in the domain of materials science and engineering, a subset of which includes corrosion science. The purpose of integration is to decrease costs and time associated with research and development. In the context of corrosion, it is proposed that informatics can produce superior decision making tools, decrease risks of failure and improve asset management. An integrated approach is necessary for corrosion because of the multiphysics nature of its contributing mechanisms that include processes at the megascale, materials deformation, electrochemical reactions and fluid dynamics. A hierarchy is introduced that combines models from these subdisciplines with models at more fundamental scientific levels (thermodynamics, microstructural, quantum mechanical and density functional theory/atomistics) and methods for treating uncertainty (Bayesian inference, Monte Carlo and reliability methods). To demonstrate the multiphysics approaches currently available for corrosion prediction, applications are drawn from the recent literature and categorised by topic: general corrosion, localised corrosion and passivity, environmentally assisted cracking, and coatings and inhibitors. Opportunities for integration in each of these subthemes are suggested. Some remarks concerning the integration of probabilistic with deterministic models are made because of the importance of attaching uncertainties to the predictions made by corrosion models, and applying a time-invariant scientific approach to the interpretation of a time-dependent historical record. Finally, a strategy for implementing the integrated approach to corrosion modelling is presented, under the name ‘corrosion informatics’.
An integrated approach to corrosion modelling
Corrosion is a spontaneous process that undermines the integrity of metallic materials used in industries ranging from health care to energy, from military to transportation. 1 The development of new materials – lighter in weight, multifunctional, or more tolerant to extremes of temperature and pressure – and methods of manufacture requires analogous developments in the models used to anticipate their response to chemical and mechanical stress. Since corrosion phenomena fundamentally consist of discrete atomistic events associated with surface chemical reactivity, and yet are controlled by mesoscale and macroscale effects such as electrochemical interactions between cathodic and anodic sites, kinetic limitations associated with mass transport, and microstructural effects in the material itself, 2 such developments must encompass a multiphysics awareness, and in some cases, require a direct multiphysics computational model.3–6 In the present review, the author considers developments in these directions for the topics of general corrosion, localised corrosion and passivity, environmentally assisted cracking and coatings and inhibitors.
Modern computation allows the modelling and simulation of complex physical processes with increasing access to arbitrarily high levels of physical rigour. As models become more complex, however, they also become more codependent. All models have inputs, these may consist of thermodynamic or kinetic parameters, component geometries, materials composition and microstructure, etc., and these inputs may be obtained either from direct characterisation, empirical fits of independent parameters to the observed values that depend upon them, or from other models – such as the use of density functional theory to build a reference library for the development of many-body potentials that can be used in the simulation of materials failure pathways.7,8 This last point leads to the possibility of multiscale modelling. 3 Although the multiscale modelling paradigm can provide several benefits, it should only be considered as a subset of a much broader vision for integration in materials science, recently encapsulated by the Materials Genome Initiative: the materials science version of informatics. 9
Informatics is defined as the ‘collection, classification, storage, retrieval and dissemination of recorded knowledge’. 10 As models proliferate in number and complexity, 11 the data used as input, the data produced as output, and the algorithms themselves need to be organised and disseminated to maximise their effectiveness. 12 The Materials Genome Initiative was created to develop the framework necessary to unleash the power of modern modelling and simulation, combined with high-resolution characterisation techniques such as atomic force microscopy (AFM) and X-ray diffraction computed tomography (DCT), to accelerate materials design and deployment. 9 Necessarily, this should incorporate a means for designing with corrosion resistance in mind and deploying materials that have accurate lifetime reliability and risk assessment models.
One of the key themes of informatics is the integration of modelling, characterisation and experiment for predicting system behaviour along with a means for quantifying uncertainty. 13 The tree diagram in Fig. 1 presents a proposed integration pathway for corrosion informatics. The hierarchy links modelling from various time and length scales as well as subdisciplines with analogous characterisation techniques and a parallel branch that lists those sources of uncertainty and methods that may be used to provide a means for placing confidence limits on the overall model predictions.
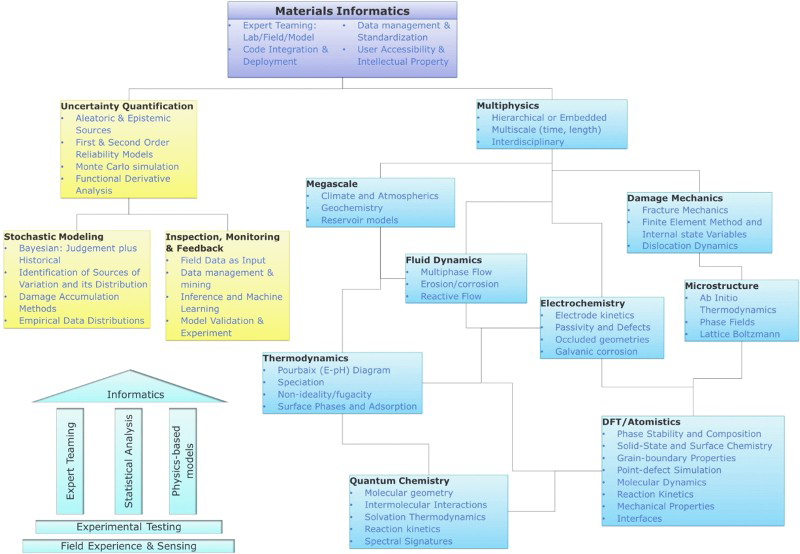
A proposed Materials Integration hierarchy with a focus on the modelling of corrosion (alternative focuses could be on advanced characterisation, not pursued in this review). On the left-hand side are the methods for quantifying uncertainty in models, in which case experiment and characterisation play a significant role. On the right are different domains of modelling. Corrosion informatics provides a unified framework for connecting these approaches. A summarised pictogram of the integration is shown in the lower left
At the top of the hierarchy are high-level informatics tasks: expert teaming between laboratory measurement and characterisation, field testing and model development, integration and deployment of modelling codes, management and standardisation of data, and accessibility of models and data for potential users while retaining appropriate intellectual property measures. An excellent example of this last point is the development of a dual strategy for deploying the open-source FREECORP corrosion model alongside the proprietary code MULTICORP by the Ohio University's Institute for Corrosion and Multiphase Technology.14,15 Data management could be addressed, for example, by the use of semantic frameworks, such as chemical markup languages. 16 The key principles of integration are summarised in the lower-left pictogram.
Uncertainty quantification (the left branch of Fig. 1) must treat aleatoric (statistical) and epistemic (knowledge) sources of error in models. 17 The former class, related to system variables, is more easily treated as statistical methods such as first- and second-order reliability methods and Monte Carlo simulation and are well developed.18,19 The latter class is more difficult to quantify; it derives from a lack of knowledge. Knowledge gaps come in several varieties; the solution to physical equations, for example, may require approximations in order to obtain a tractable solution; more profoundly, models only describe a subset of possible physical mechanisms that can cause corrosion, yet there may be many others, both known and unknown, that are not given consideration. When solutions of competing rigour are available, the functional derivative method may be applied to estimate the uncertainty introduced by using faster, more approximate solvers. 20
Models that explicitly address uncertainty include Bayesian inference, in which expert judgement (including empirically derived heuristic knowledge and science-based models) is used to form a prior probability distribution that is then updated with historical and field statistics. 21 Scientific and statistical models can both benefit from a consideration of the sources of variation. For instance, the susceptibility to stress-corrosion cracking (SCC) may be dependent upon the statistical distribution of certain grain boundaries within the material microstructure.22–27 Thus, a science-based model for SCC could benefit from including a distribution of crack propagation parameters, depending on the types of grain boundaries in a given material, rather than treating the parameters as single valued. Deterministic and probabilistic effects can also be reconciled through damage accumulation functions. 28 In some cases, corrosion phenomena have also been treated from a purely stochastic standpoint, using extreme value statistics generated empirically. 29
Ultimately, the confidence placed in a model derives from the ability for a model's predictions to compare with ‘real data’, obtained from the laboratory or the field. This ‘litmus test’ comprises the Inspection, Monitoring and Feedback module of Fig. 1. Field data should be used in two capacities: as input to the models, but also as a check against models, especially in their early phase of development. As sensor technology continues to improve, data management and mining practices will need to develop alongside. Inference and machine learning techniques should process data in an intelligent way by integrating data with models. Finally, the comparison of a model with experiment and/or field data will remain the most important test of a given model's utility and reliability. It should be remarked that, in some instances, models may be useful without being especially accurate when they advance understanding or are suggestive of new practices or directions of inquiry. 30
Since the consideration of uncertainty applies to a variety of models, they have been considered distinctly from the modelling paradigms presented on the right side of Fig. 1. The modelling paradigms will now be given consideration.
Multiphysics methods involve the coupling of models for the purpose of simulating connected phenomena. For instance, diffusion of hydrogen into a material may be modelled in parallel with the evolution of mechanical processes to provide an overall description of environmentally assisted cracking. 31 Additionally, one might also incorporate an atomistic description of the interaction between hydrogen and dislocation cores to derive from first principles the influence of hydrogen on the cohesion zone mechanics. 32 Multiphysics models may be hierarchical – one physics model produces parameters that will be inputs for another model – or embedded – in which case the two models are ingeniously reconciled within a single algorithm or set of coupled equations. 3 The advantage of multiphysics methods is that high levels of physical rigour can be selectively applied only where needed, as increasing rigour typically translates to increasing computational cost and complexity. This advantage allows simulations to bridge greater time and length scales. Multiphysics models are, by definition, interdisciplinary, underscoring the need for an integrated approach that utilises teams of experts across the science and engineering disciplines.
Within the scope of this review, four fields of inquiry are identified as categories from which multiphysics models may be drawn: megascale techniques, fluid dynamics, electrochemistry, and damage mechanics. Megascale methods apply on global length scales: climate and atmospheric modelling, geochemical models and reservoir modelling fall into this category. 5 Fluid dynamics models can predict the dynamics of multiphase flow, and thus stresses and other parameters relevant to erosion/corrosion and reactive flow.33,34 Electrochemical methods are foundational to aqueous corrosion and include electrochemical kinetics, models for passivity, such as the point defect model, mass transport models that treat occluded geometries such as cracks, crevices and pits, and Galvanic corrosion.35–38 Damage mechanics methods include models based on fracture mechanics, direct simulation of solids via the finite element method and internal state variables (ISVs), and dislocation dynamics.6,26,39–41
To provide fundamental information necessary for the solution of engineering scale models, a number of science-based models are required. These include thermodynamic methods, such as Pourbaix diagrams, speciation models, nonideality equations and surface stability models42–46; quantum chemistry, for the prediction of molecular structure and properties, including intermolecular interactions, solvation thermodynamics, reaction kinetics and spectral signatures47–49; density functional theory and atomistic simulation, for interrogating the relative stability of different phases, solid-state and surface chemical reactions, properties of grain boundaries and point defects, performing molecular dynamics simulations, predicting reaction kinetics, mechanical properties and identification of the structure, composition and stability of interfaces38,50–54; finally, the authors consider microstructure-based approaches, such as ab initio thermodynamics, phase field theory and lattice Boltzmann methods.40,55 As indicated in Fig. 1, there are many possible ways that the science-based methods can inform and supplement the engineering scale models, as well as one another.
Informatics in materials and chemical engineering
Since concepts associated with materials informatics are still being defined, it is helpful to clarify by considering some analogies in materials science and chemical and aerospace engineering. For an example in the world of materials science and engineering, consider the development of ‘cybersteels’. 56 Cybersteels, according to Olson's definition, are ‘hierarchically structured multiphase, multicomponent materials’ designed according to the principles of materials informatics (which he calls genomics): i.e. the integration of ‘materials science, continuum mechanics and quantum physics… theory, simulation and experiment’ to develop ‘process/structure/property/performance relations’, which enabled designers to select optimal composition and processing parameters. Consequently, the materials were developed and deployed in a rapidly reduced timeframe, compared to traditional metallurgical approaches, with significant cost-savings. A pivotal piece of this work involved the calculation of phase diagrams (CALPHAD) from ab initio data. 57 3D characterisation tools – FIB, microstructural tomography – provided background information for ‘numerical simulators’ to candidates of mine materials that met the requirements for ‘strength, toughness and fatigue resistance’. Methods were developed to predict precipitation strengthening and particle size from elastic energies and multiscale modelling. Intergranular embrittlement was minimised by density functional theory calculations of interface properties and Rice–Wang thermodynamic theory. 58 Following these principles, a ‘genomic fundamental surface thermodynamic’ database was created to ascertain the boundary conditions that controlled the effect of hydrogen, and to then create a materials microstructure that ‘completely eliminated’ the possibility of intergranular SCC. Whereas expert guidance, scientific principles and the feedback between metallurgical engineering and materials characterisation have always been a part of the design cycle, for example, the use of Ashby diagrams to guide materials selection, 59 it is the integrated approach – in particular, the integration of computational tools that are now for the first time of sufficient accuracy and usefulness – that is of particular significance in accelerating the process. Before this era, theory, modelling and simulation were playing ‘catch-up’ to the advances in characterisation and experimentation in materials science; in the materials informatics age, the methods are now on par, with the relative ease, cost advantages and speed of computational modelling now even beginning to take the lead over conventional Edisonian lab-bench practice.
An informatics approach has also been advocated in chemical engineering. The production of chemicals requires a consideration of molecular properties that constitute the raw materials as well as the final product, the processes that transform those molecules throughout a production process, and then the large-scale issues that define process engineering.60,61 Analogous branches to the construction of Fig. 1 apply to chemical manufacturing. The pressures driving innovation in chemical engineering are similar to those in materials – market demand for new products, public concerns over safety, security and the environment, and a competitive climate that requires a rapid product design cycle. The conversion of mass and energy into products via chemical engineering has its doppelganger in the conversion of metals into corrosion products and entropy during environmental degradation. The processes considered by a chemical engineer must encompass timescales from pico- and nanoseconds of chemical reactions up to hours for operating industrial processes and centuries for pollutant destruction as well as length scales that must span nanoscale (10− 8m) to microscale (particles, eddies) to macroscale production units (cm-m) and megascale for dispersion of emissions (km-1000 km), in direct analogy to the multiscale nature of corrosion. An example of the use of multiscale methods that incorporate quantum mechanics for facilitating product safety assessments was given by Lewis et al.
62
The informatics approach to chemical engineering and model development was summarised by Charpentier as follows:
“translating molecular processes into phenomenological macroscopic laws to create and control the required end-use properties and functionality of products manufactured by a continuous process. [coupling] thermodynamics, kinetics, rheology and transport … toward the conception of integrated operations… Modeling must be an activity that requires knowledge and comprehension of scientific facts, experience, skills and judgment.”
57
A bold vision for the application of a truly integrated approach to asset management, of which corrosion prediction and control form a subset, is in the ‘digital twin’ aerospace concept proposed by Tuegel et al. 63 Derived from the perspective of the US Air Force, the Digital Twin imagines a future capability in which computational models simulate (with ultrahigh fidelity) a 1 h flight using only 1 h of computation time, and this simulation is used to estimate the accumulated usage damage, the probability of mission success, and the probability distribution for the remaining useful lifetime for the aircraft. Alongside this simulation technology are similarly developed sensor technologies in the vehicles themselves, thus creating the digital twin, in which the sensor could provide new inputs to the models – thus keeping them up to date in real time – but also feedback to improve the models and capture uncertainties. This vision that would require very high-performance digital computing – ‘exascale computation’ and ‘petabyte’ data storage – is multidecadal, requiring advances in not only hardware but also in conceptual approaches to algorithm development and multiphysics/multiscale software architecture. One can conceive that this ‘digital twin’ concept could be applied to structural life prediction across industries beyond defence. This generalisation is consistent with the definition of the digital twin as provided by Stargel and Glaessgen: ‘an integrated multiphysics, multiscale, probabilistic simulation of an as-built vehicle or system that uses the best-available physical models, sensor updates, fleet history, etc. to mirror the life’ of its real-world counterpart. 64 The method already has a pre-existing parallel with the Multi Analytic Risk Visualizaton tool (MARVTM), which is a Bayesian network tool developed for oil and gas pipeline risk assessment.65–67
The goal of ultrahigh fidelity, such that the Digital Twin would be practical and useful, requires several revolutionary advances in the following key areas:
multiphysics modelling multiscale damage modelling integration of structural and damage models uncertainty quantification and management the manipulation of large datasets – ‘big data’ high-resolution structural analysis.
These areas for improvement will also apply to the ultrahigh fidelity modelling of structural materials in other industries. For instance, the integration of structural and damage models involves the coupling of physics models for materials microstructure evolution and defect accumulation with finite element models for the structural response to gradients in temperature or applied stress, such as that occurring in fatigue cracking. Probabilistic estimates of a statistically distributed microstructure with statistically distributed defects can be augmented by known defect distributions obtained through sensor measurements.
The proper handling of uncertainty is critical to the digital twin concept. If uncertainty is large, there is no point spending CPU cycles computing physics-based models at ultrahigh fidelity. Rather, the appropriate level of rigour should be selected based upon the uncertainty of the input data and model parameters. Tuegel et al. point to stochastic finite element modelling as a path forward in this regard, and consider alternative approaches such as Monte Carlo sampling to be too time-consuming and costly for the generation of overly detailed probability distributions from more widely uncertain data. 63
Corrosion: Recent modelling developments
The preceding sections promote an integrated approach to corrosion modelling that balances science-based models with engineering models and approaches to treating uncertainty, while also incorporating expert judgement and experimental/field data. Recent developments that provide waypoints towards this destination will now be summarised across the major corrosion subtopics of general corrosion, localised corrosion, environmentally assisted cracking and coatings and inhibitors.
General corrosion
In this section, fundamental models that address general aspects of corrosion are considered: simulations of surface chemistry, diffusion and electron transfer.
The study of the fundamental molecular processes associated with corrosion has progressed alongside the development of more efficient computational methods for solving the electronic structure processes associated with the changes that occur in local bonding and coordination at the metal/electrolyte interface. 68 Early efforts to simulate the processes of dissolution and oxidation at the iron/aqueous interface were made by Anderson and colleagues in the early 1980s.69,70 A molecular orbital theory method was used to simulate the changes in coordination of Fe atoms in the presence of adsorbed OH. More recent innovations include the use of density functional theory to simulate the presence of ordered states of aqueous solution on metal surfaces, including the surface induced dissociation of water to form hydroxide and/or oxide two-dimensional phases precursors to passivity.71–75
An effort combining molecular dynamics and constrained geometry optimisation was made to simulate the process by which Cu adatoms dissolve from a nanoparticle surface facet.76,77 The change in potential energy of the dissolving ion, as a function of its progress across the electrochemical double layer was shown to be highly dependent on the extent to which the coordination sphere of water molecules was allowed to relax. Further investigations of this nature are required in order to be able to directly compute activation barriers and transfer coefficients for the anodic dissolution reaction.
The dependence of adsorbed metal atom potential energies on the local structure and composition was characterised using both density functional theory and interatomic potentials, such as the modified embedded atom method.76,78 In some cases, simulations using interatomic potentials have been used to simulate both oxidation and electrochemical dissolution of metallic surfaces.79–81 The validation of such simulations, however, requires continuing high-resolution characterisation efforts. Quantifying the errors associated with the molecular dynamics simulations – usually performed over very short timescales – and those errors introduced by the use of approximate interatomic potentials is challenging, and an active area of research in the computational materials science community. 82
Despite the challenges associated with capturing the uncertainty in these models, interatomic potentials have been used to perform kinetic Monte Carlo simulations of alloy dissolution, as a means to discern the general structure-property relationships that control the stability of a given alloy matrix to corrosion. 83 For instance, it was demonstrated, for Tc–Fe alloys, that variations in the Tc content could be used to minimise the vulnerability of the Fe matrix to dissolution. 84 Grand canonical Monte Carlo simulations have been performed in a similar way to illustrate the factors controlling crystallographic pitting of hexagonally close packed metals.36,85 More sophisticated models of this variety need to be developed that incorporate additional chemistry and solid-state reactions, including the formation of oxide films, the interactions between anodic and cathodic sites in the material, and the role of solution phase species – water, ions, hydrolysis products – in controlling the dissolution reaction kinetics.
Investigations of binary alloy dissolution have shown that two phenomena are critical for determining overall system behaviour, while dissolution is the most obvious, the other phenomenon is surface diffusion. 86 It was shown, for instance, by in situ scanning tunnelling microscopy that dissolution of copper is mediated first by the diffusion of Cu adatoms away from the step edge and onto a terrace site, where its coordination to other metal atoms (and hence surface stability) is greatly reduced. 87 A detailed electrochemical model for the diffusion properties of metal surfaces, including the effects of surface heterogeneities (steps, kinks), electrochemical fields and adsorbed anions (chloride) was recently developed based on a multiphysics approach that combined density functional theory, interatomic potentials, electrochemical theory and kinetic Monte Carlo simulation.88,89
Since aqueous corrosion is electrochemical in nature, it is also pertinent to consider the reactions associated with the cathodic site: oxygen and hydrogen reduction.88,90–96 The quantum mechanical basis for electron transfer was laid out by Levich. 97 Intrinsic to this theory is the necessity of computing coupling constants between the orbitals of the two species undergoing electron transfer. In cases like hydrogen reduction, the interaction of the electron acceptor orbital with the band structure of the electrode defines this coupling constant and can be determined from electronic structure models, like density functional theory. The mediation of this reaction by the adsorbed state of hydrogen (in the Volmer–Heyrovsky mechanism) was studied by Schmickler et al. and the significance of the structure and proximity of the metal d-band to the Fermi level shown to be a defining factor.94–96,98,99 An extension of this approach has also been made to study the electron transfer mechanism of anodic dissolution and its reverse process, deposition. 100
Electrochemical models for simulating general corrosion exist in various stages of sophistication. These models are almost entirely empirical in nature and based upon general principles of electrochemistry. A recent innovation couples a finite element simulation of the part geometry and material composition to predict galvanic corrosion of component assemblies.35,101 The inputs to the model are polarisation curves, obtained from experiments on the materials pertinent to the assembly design. The outputs are corrosion currents as a function of position along the input component structure. Geometries can be optimised, materials selection made, and coatings decided upon, in addition to computing corrosion currents as a function of position.
Convergence of these two classes of models – discrete, atomistic simulations with continuum, numerical solution of analytical electrochemical equations – will take place as the atomistic simulations expand to take into account more complex surface geometries, involving defects and microstructural effects, and, at the same time, the continuum solutions move into finer and finer mesh sizes, and seek to describe scenarios that approach the same mesoscopic level (see Fig. 2). At the same time, this convergence is unlikely to arise naturally: a focused and interdisciplinary effort will be required to develop simulation methods that are robust enough to handle the complexity associated with corrosion phenomena, as the natural timescales for molecular dynamics simulations (femtoseconds) are six orders of magnitude lower than the timescales required to simulate the phenomena associated with anodic dissolution. 102
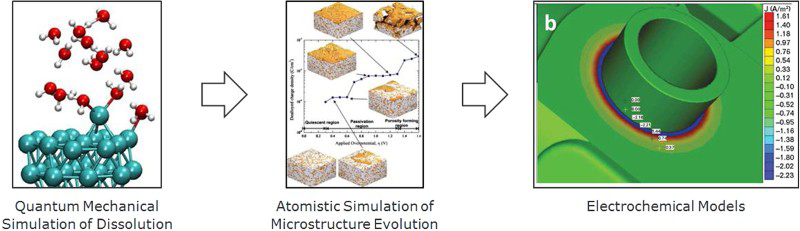
Example integration pathway in which fundamental modelling at the quantum mechanical level can guide atomistic simulations of microstructure/corrosion relations that ultimately inform continuum scale electrochemical models for materials corrosion.
Passivity and localised corrosion
The presence of oxide films, which have thicknesses ranging from one to hundreds of nanometres, generally provides a material with resistance to corrosion, particularly at low and ambient temperatures. 103 The properties of these oxide films depend upon the material composition, microstructure, surface treatments, and the chemical environment. The development of design principles for manipulating these parameters to convey superior corrosion resistance is continuing. The integrated approach of modelling and high-level characterisation provides a strong foundation for improving upon current state of the art. In this section, recently published techniques for modelling oxide film formation and properties are described, along with models for localised degradation modes such as pitting and crevice corrosion. The models span the categories from Fig. 1 of density functional theory and atomistics, thermodynamics, microstructure and electrochemistry.
Oxide formation on metal surfaces can be templated by the early process of oxygen chemisorption and then reconstruction of the chemisorbed phases upon the growth of the oxide film. The transition from chemisorbed layers to the first few layers of oxide on nickel was recently examined for close packed and stepped surfaces using density functional theory. 104 Some fundamental questions were explored, such as the nature of the electron transfer from nickel to oxygen and the progression in oxidation energy from chemisorption up to multilayer formation. Similar studies have been performed on Mg(0001) and Pt(111).105,106 The application of the first principles parameters into an ab initio thermodynamics framework enables connections to be drawn from the quantum chemical observables (like binding energy) to the macroscopic observables, such as two-dimensional surface phases. These studies, although confined to only a few atomically thick layers, also provide a foundation for the studies of oxidation kinetics, the role of alloying elements in potentially controlling the oxidation process, and large-scale molecular dynamics simulations of oxidation and related phenomena (such as passivity breakdown) using interatomic potentials.
A number of molecular dynamics simulations of this nature have been reported, for systems such as nickel, iron, aluminium and zirconium.79,107,108 The development of ‘reactive force fields’ has allowed a new generation of molecular dynamics simulations to be performed that can take into account not only changes in local geometry of atoms but also changes in their oxidation state, which typically lead to more drastic changes in the properties of the material.109,110 These simulations have been used to examine the mechanisms of oxide growth (as mediated by point defects), and the effects of temperature and surface orientation. As these models improve, based upon the development of more reliable potentials, they will have the potential for integration with electrochemical models for passivity. For example, mesoscale models developed using cellular automata have been used to address the relationship between adsorption of thin oxide films and the formation of thicker passive layers, and their significance to the interpretation of polarisation curves, 111 as well as simulation of chloride-induced pitting of bimetallic materials, 112 and pitting morphology. 113
The point defect model provides an electrochemically based, analytical, deterministic approach to the modelling of corrosion, founded on the premise that overall corrosion rates of materials are controlled by the transport of defects across the oxide film. 37 The critical processes include the generation and annihilation of cation and oxide vacancies at the appropriate metal/film and film/solution interfaces, and the flux of oxygen and cation vacancies across the oxide film. Bilayer films could also be considered by a variant of this model. The solution to the mass transport equations via steady-state theory provides the ability to predict overall corrosion behaviour once fundamental rate constants for diffusion and transport across the various interfaces (metal/film, film/solution) are identified. Thus, the model can either be empirically solved by obtaining the rate constants from an integrated experimental/modelling program, or from low-level first principles atomistic models such as those described above.
The study of passivity breakdown has also seen treatment via a number of modelling techniques. An investigation via density functional theory of the α-Cr2O3(0001) surface enabled the first principles derivation of the adsorption energies for various environmental contaminants, including hydrogen, chlorine and sulphur, in addition to determining the response of the oxide film surface to the adsorption effect via geometric reconstruction. 114 A similar approach was used to examine the role of chloride in changing the surface structure of Ni(OH)2, first using density functional theory, 115 and then, later by atomistic simulation. 116 Key challenges with these works, however, are that (a) it is nontrivial to validate the models, both from the standpoints of epistemic sources of error (simulation methodology) and the paucity of sufficient high-resolution surface science data to provide a direct side-by-side comparison, and (b) that the results then need to be integrated into a multiphysics approach that connects the findings of the interatomic potential simulations with macroscopic parameters, such as corrosion rates or other electrochemical observables. One effort to move towards the modelling of kinetics is the virtual lattice model developed for Fe–Cr alloys, in which the formation of chromium oxides over an iron-based alloy is simulated using kinetic Monte Carlo. 52
An innovative approach that combined fundamental surface science theory with electrochemical models and thermodynamics was used to predict the threshold potentials for localised corrosion using the repassivation potential as a lower bound for the pitting potential.46,117 By breaking down the processes that compete for surface activity – including dissolution, chloride adsorption, oxygen chemisorption, and, in a more recent model, sulphide adsorption – the potential required to repassivate the surface was solved through steady state theory. The fundamental parameters for the model, including adsorption energies and activation barriers, were determined in the original implementation by fitting the model to experimentally determined repassivation potentials, although one can also conceive the use of more rigorous physics-based models, such as density functional theory, to provide those model inputs. Integrating density functional theory with surface thermodynamics enabled the prediction of boundary conditions for the ammonium chloride corrosion of mild steel. 118
The breakdown of passivity is typically associated with localised corrosion: fluctuations in the oxide film properties because of defects or underlying fluctuations in the materials microstructure may create vulnerabilities that are spatially associated, or, conversely, mechanical or environmental effects may create local breaches in the oxide film because of physical damage (oxide film fracture) or occluded conditions (crevices), which chemically destabilise the oxide film. In such cases, localised corrosion will only be sustained as long as the environmental conditions persist. Since aqueous corrosion requires the transport of ions between cathodic and anodic sites, the continuation of corrosion will therefore depend upon the nature of the mass transport. With this in mind, there has developed a body of electrochemical models that simulate the progress of corrosion in such localised systems using reactive transport models, in which case the corroding surface and the external (bulk) environment provide the electrochemical boundary conditions and Fick's law is used to solve the diffusivity problem, thus predicting the extent to which localised corrosion (pitting or crevice corrosion) develops over time.119,120 Such reactive transport models have been used, for example, to simulate the corrosion of spent nuclear fuel assemblies over time 121 and to evaluate the corrosion mechanisms for H2S corrosion of stainless steel. 122
The simplest such models simulate the electrochemical reactions and transport with a one-dimensional finite-difference model. Electrochemical measurements can be used to inform the model parameters when fundamental information is not available. For example, a one-dimensional model for the transport, reactions and variation in electrochemical potential was used to model a four component alloy (Fe–Cr–Ni–Mo, i.e. 316L) via the technique of finite difference. 123 A critical pH was used as the reference point at which value the crevice moves from passive to active corrosion. Changes in the pH were made according to hydrolysis reactions in the model. In an integrated approach, these reactions could be determined from quantum chemical simulations, 124 just as the rates of surface reactions could be determined from first principle density functional theory calculations and the role of microstructure incorporated, rather than treated as a continuum bulk substance. The role of crevice width: length ratio was investigated to identify the critical geometry required for the initiation of crevice corrosion.
Transport models have also been used to simulate the localised corrosion of steel reinforcement within concrete structures. A systematic approach was developed to simulate, using a combination of chemical, thermal, and mechanical models, the ‘complex non-mechanical and mechanical processes in concrete’ that control the corrosion of reinforcing steel.125,126 The first stage of the model addressed depassivation via a threshold concentration model: a finite element model was developed to model the mechanical damage of the concrete, the transport of water and chloride into the concrete medium and its diffusion through cracks. This was the first step towards a model that can ‘realistically simulate corrosion of reinforcement, before and after depassivation of steel, for any geometry, boundary conditions and loading.’ The model depends on a number of parameters: moduli of elasticity of concrete and steel, porosity, heat capacities, hydration time constants, chloride diffusion activation energies, etc. that could be determined from lower length scale models, or be accessed from a database depending on material type. The depassivation process was not explicitly modelled but assumed to occur once the mass concentration of free chloride in pore water reaches the empirically determined threshold value of 7 kg m− 3. To model the formation of corrosion product (which would lead to cracking), the authors then developed a three-dimensional model that computed the corrosion current density as a function of water saturation and three-dimensional geometry, by finite element simulation of a macrocell for depassivated steel embedded within concrete. Effective diffusion coefficients were used for the transport of oxygen through the concrete. The steel exposed to a crack was assumed to be the anode with the cathode supplied by the unexposed portion of the metal (which has access to oxygen via the diffusion through the partially saturated concrete). Exchange current densities, equilibrium potentials and Tafel slopes were used as input – hence testing and empiricism must supply those numbers, lacking a first principles derivation. The pre-existence of some crack geometry was also required to be input for the problem setup. As seen in the following section on environmentally assisted cracking, emerging methods in tomography and microstructural simulation will lead to future models that directly simulate crack morphologies. Similar finite element methods coupled with reactive transport equations have been used to explore short and long term issues related to the crevice corrosion of corrosion-resistant alloys in biological media. 127
The integration of models from the atomic scale up to the megascale was explicitly addressed in the review paper by Gunasegaram et al.
4
The authors, in particular, identified knowledge gaps that would need to be addressed to facilitate an integrated approach to provide practical and powerful computational tools for predicting and then designing against localised corrosion. In the case of most design models, materials are typically modelled from a homogeneous standpoint, and this is considered to be the primary way in which materials models at lower length scales can contribute by identifying the ways in which materials heterogeneities (in the underlying material and the oxide film) contribute to variations in localised corrosion. Other challenges include the integration itself – how to separate out the various scales and assign methods appropriately, improvements in the efficiency of each of the separate modules being integrated such that the simulations can be performed accurately yet in a timely manner, linking the models, and, finally, the validation. The authors proposed a multiscale modelling strategy that decomposed the localised corrosion problem into the following processes:
metal oxidation metal ion dissolution, diffusion, speciation and hydrolysis oxygen reduction OH-ion dissolution, diffusion and further solution reactions precipitation of corrosion products effect of initial oxide layer solution chemistry (migration and diffusion).
As reviewed both in the present paper and by Gunasegaram et al.,
4
various pieces of this overall process have been modelled to greater or lesser extents, thus indicating that the trajectory towards such an integrated approach to a complicated materials failure mode (i.e. localised corrosion) has already begun.
Environmentally assisted cracking
Owing to the interdependence between chemical environments, materials microstructure, mechanical effects and apparently stochastic behaviour, prediction of materials failure via environmentally assisted cracking, like localised corrosion, is a challenging field that requires a multiphysics approach. Furthermore, the environmental and microstructural conditions that can result in cracking may have different incubation times, which means that failure modes may not emerge during accelerated testing and, lacking a multifaceted modelling strategy, appear entirely unpredictable. This challenge was recognised by Staehle in the microprocess-centric approach, which divides the challenge of predicting environmentally assisted cracking into domains and microprocesses. 128 By categorising the phenomena that lead to the accumulation of corrosive behaviour and changes in defect structure of the material across the domains of the global system – the bulk environment, near-surface environment, the passive film, the near-surface metal and the bulk metal – a systematic framework was established for predicting failures that have not yet occurred (see Fig. 3). Such a framework requires the integration of high-level characterisation tools, systems monitoring, experts across the fields of chemistry and materials science, experimental testing and high-fidelity modelling from the atomic scale to the systems level. Examples were provided in the original paper for how this approach could be used to predict sulphur and lead-assisted SCC in a light water reactor environment. 128 A direct analogy to this multiscale viewpoint can be seen in the GILDES model for atmospheric corrosion constructed by Graedel, where GILDES is defined by the domains of gas, interface, liquid, deposition layer, electrodic regime and solid.129,130

Illustration of the decomposition of a system that is vulnerable to environmentally assisted cracking via the domains–microprocesses sequence approach 128
Owing to the difficulties of reconciling chemical and mechanical effects within a single model, most modelling works that treat environmentally assisted cracking focus on either one effect or the other. Industry models have been developed that can predict allowable stress thresholds for a given size of corrosion induced defect, and these have been evaluated by finite element models that reconcile the mechanical pressures leading to failure, and the change in corrosion activity around the stressed defect. 131 Finite element approximation models of this type require specific materials and environment inputs that must be determined empirically or from models that incorporate multiphysics effects. As output, they establish relationships between the tolerable defect size and maximum internal pressure under conditions of soil-induced strain and corrosion. The advantage of finite element analyses is that they can be readily applied to defect structures obtained from direct observation of mechanical or structural components. For example, dents characterised via inline inspection technologies have been used as input geometries for finite element elastic–plastic models to assess the susceptibility of physical dents to initiate cracks. 132
The use of ‘ISVs’ is critical to the application of finite element models for simulating the effects of damage, history and plasticity and to represent the growth of, for example, voids, slip-systems and dislocation densities. The development of ISV theory and its implementation in finite element simulations of materials is reviewed by Horstemeyer and Bammann, 133 who mention that the current state of the art allows the prediction of structure–property relations at such a high level that now it is possible for machine component materials to be optimised (an example was given of a Cadillac control arm that was reduced in weight by 25%, cost by 12% and, at the same time, increased in load-bearing capacity by 50% and fatigue life by 100%). 133 In such an approach, it is necessary to specify the appropriate thermodynamic constraints (energy, momentum conservation and third law restrictions), as well as requiring only physically admissible models (i.e. models that ‘identify the physical mechanism or discrete microstructural feature at the particular length scale that is a root source of the phenomenological behaviour’). Internal state variables should be associated with the different length scales associated with the mechanisms of deformation (lattice parameters for atomistics, Burger's vector for dislocation stress, diffusion to grain size, dislocation densities with second-phase particles, …). Consistent with the ISV philosophy, an abstract framework was developed for introducing the damage induced by uniform, localised and intergranular corrosion into the finite element model, although the exact details for how the chemical effects modify the mechanical response have yet to be worked out. 41
Recent developments in the finite element modelling of materials failure via cracking have enabled the introduction of microstructural effects, down to the role of grain boundaries via the introduction of interface elements (elements of zero thickness that represent the crack front region). de Borst reviewed the challenges in computational materials science arising from the need to incorporate discontinuities into continuum finite element simulations because of grain boundaries in crystalline materials, solid–solid phase boundaries and discrete dislocations, among other microscopic and submicroscopic aspects. 3
Advances in characterisation of crack pathways have allowed models to be built that directly include information regarding the orientations of grain boundaries that fail because of intergranular cracking. A probabilistic model that includes such information was recently derived. 39 The link between grain shape, grain size, grain boundary orientation and crack propagation behaviour/percolation parameters was investigated using a Markov chain technique with ‘realistic’ grain structure created using a Voronoi model with grain centres distributed according to the shape parameters of the real material [obtained from electron backscatter diffraction (EBSD)]. The simulation of the cracking failure was two-dimensional, and the role of the environment was not taken into account, other than implicitly, through the information gained empirically for cracking susceptibilities. Cracking probabilities for the Markov Chain model were distributed according to the distributions of grain boundaries in the material [i.e. low angle, high angle and coincident site lattice (CSL)], with the probability of cracking for each category given from the observed crack pathways in actual experiments. The model itself, therefore, was not physically based, but could only be applied to the particular material and environment that the experiments were performed on. At the same time, the model illustrates a pathway to incorporate materials microstructure effects into models for environmentally assisted cracking.
Along similar lines, a finite element model was constructed using fundamental microstructural information obtained via X-ray diffraction contrast tomography. 6 The model resolution was down to grain sizes on the order of tens of micrometres. The orientation relationships between the grains were used to estimate susceptibility similar to the above rankings used in the two-dimensional model just reviewed. Coincident site lattice and low-angle grain boundaries were considered to be less susceptible to cracking. An approximate piecewise-linear traction curve was used to simulate the separation. The model contained no chemistry or materials defects, other than the grain boundaries. The model represents a step-forward in the state of the art simulation of materials fracture in that intergranular cracking was simulated with realistic microstructure. The inclusion of modified traction curves, because of chemical effects on grain boundaries, or dislocation dynamics would represent the next step in multiphysics modelling of environmentally assisted cracking. 134
Interactions among microstructures that develop in multiphase steels were simulated using finite element method and the use of representative volume elements for the contributions made by each of the various phases that contribute to the steel microstructure. 135 Von Mises elastoplastic relations were assumed for each single phase. Two approaches were numerically investigated: failure by porosity induced by the inclusions, and failure induced by the austenite–martensite transformation, which takes place at high strains. In this model, interface elements were used to simulate cleavage fracture. It was acknowledged that the use of interface elements can artificially constrain the simulation of a cracking trajectory; in that crack, paths must be assumed from the beginning.
One model that combines mechanical and electrochemical effects is based on the slip-dissolution theory, which states that fracture of the oxide film via stress allows a localised corrosion event. Repassivation then competes with crack growth. The tension between crack blunting (via corrosion) and crack propagation (from the mechanical forces) is used to define the criterion for SCC. This model was applied to sour gas service conditions. 136 Slip-dissolution theory was used to predict the minimum time a pit must survive (before repassivation) to allow cracking to occur. The Kondo–Tsujikawa criteria were used in combination with the Cogleston correlation to estimate this minimum time. The analytical expressions derived use the slip-dissolution model, and therefore include parameters for the oxide films strain resistance, mechanical properties of the material, and the K IEAC for the material. Using parameters derived for 300 series stainless steel, the key limiting factor was applied by the Kondo criterion, that is, in terms of a minimum stress-intensity factor, rather than the Tsujikawa criterion, that is, the crack must grow faster than the corrosion pit. By examining transients, the authors analysed the probability of cracking and the expected time to failure, from a Poisson distribution for the repassivation time. The two key results obtained from their model included the likelihood of cracking and the forecast time to failure, derived from the above criteria, and measurement of the pit current density and the pitting event frequency.
Since the processes that lead to environmentally assisted cracking are fundamental molecular (in the environment) and then atomistic (as the environmentally introduced impurities interact with the material microstructure) an emerging class of materials simulations have attempted to reconcile the atomistic aspects with the continuum stress-strain relations of the material. As summarised by Curtin and Miller:
“… an ideal goal is to simulate the behavior of materials with the only constitutive law being the interactions between the atoms. But due to the scales of deformation that are important in realistic problems, explicit model of all of the atomic degrees of freedom will never be feasible. Thus, one must selectively remove most of the degrees of freedom to make the problem tractable. The vehicle for this reduction of degrees of freedom is the continuum, where for crystalline materials the atomic positions can be tracked via displacement fields interpolated between a sparse set of nodes.”
137
The technologies underlying such continuum/atomistic linkages have been reviewed by Brenner. 138 A multiphysics approach entails challenges, such as dealing with the artificial forces introduced by incomplete bonding at the atom/continuum interface, phonon dynamics and heat transfer (since finite element nodes can not displace at the same wavelength as atomistic nodes, and heat transfer is also a problem for interatomic potentials that do not include electronic conduction), and the ‘reverse mapping problem’ of going from finite element nodes to create a representative atomistic environment (because of multiple possible atomistic scenarios corresponding to a given finite element node state). Increasing the time domain in the atomistic simulation component poses a significant challenge, as it is this module that is the principle bottleneck because of the large number of atoms (and hence computer time) necessary to simulate extended defects in materials.
An alternative approach to bridging atomistic phenomena and the continuum is provided by phase field modelling. 55 Phase field models capture discontinuities and interfaces by diffuse interfaces – the variation of a ‘phase field’ throughout the continuum of a material. The structure and dynamics of defects are investigated by optimisation of the ‘phase field’ free energy. Dislocation cores can be treated via a phase field, as well as interfaces that include misfit, crystallisation energies, solidification, phase-transformations, and magnetisation phenomena (domains). The natural length scale of this model is nanometres. Two kinds of phase field model have been predominantly applied: microscopic phase field (MPF) and coarse-grained phase field (CGPF). Microscopic methods explore defects and microstructural features at the 1-100 nm dimension. Ab initio calculations can be used to generate information for MPF. Coarse-grained phase field is more suited for collective behaviours of microstructural ensembles – grain growth, domain coarsening, dislocation networks – the individual defects, however, get ‘lost’ in the diffuse interface treatment as these length scales are larger. Microscopic phase field calculations can be used to feed inputs to CGPF. 55 Phase field modelling can be used to model fracture and the evolution of the cohesive zone, 139 as well as crack propagation. 40 Phase-transformations (displacive–diffusive) have also been modelled with MPF and CGPF. 140 A phase field crystal (PFC) method was developed, which is physically on the length scale of atoms, but can be implemented on diffusive (10s of nanoseconds) or vibrational timescales (picoseconds). 141 These methods have yet to be more widely applied in corrosion. 142
The field of environmentally assisted cracking that has seen the most investigation from an integrated multiphysics approach is the topic of hydrogen embrittlement. Through a connected series of studies, operating at distinct length scales, the role of stress in accommodating interstitial hydrogen was ascertained via density functional theory, as was the enhanced diffusion of hydrogen across material surfaces. 143 Atomistic models have also been used to simulate the adsorption of hydrogen at materials vacancies, thus lowering the thermodynamic barrier to creating microvoids with high local hydrogen concentrations. 144 Such processes indicate the potential for impurities, particularly hydrogen, to create precursors that would lead to SCC, a framework that is inline with the philosophy of domains and microprocesses. 128 Using dislocation theory, it was also demonstrated that segregated hydrogen at impurities by the crack tip would facilitate dislocation generation and other defect modes contributing to enhanced plasticity. 32 Atomistic studies have been employed to study the interaction of hydrogen with materials features, although they have rarely been reconciled into an overall multiphysics, multiscale model.145,146 Even still, scale up to engineering level systems has yet to be obtained. Steps towards this goal will involve, along the way, including electrochemical effects, such as aqueous solution, mass transport down crack tips and occluded environments, pH, potential and temperature.147,148
Coatings and inhibitors
The native resistance of a material to corrosion can be improved by chemical or physical barriers introduced by inhibitors or coatings, respectively. The physical, chemical and electrochemical properties of these barriers and the method and frequency of their application remain topics for optimisation via a variety of modelling and experimental approaches. 149 Furthermore, the degree of protection and the mode of corrosion mitigation provided are not always easy to infer, hence there remains scope for further development within an integrated approach.
A large body of work has emerged that tackles the problem of inhibitor design by following a parallel to the use of molecular modelling in drug design. 47 Beginning in the 1970s, quantum chemical models were developed for molecular quantitative structure–property relationships (QSPR) [sometimes known as Quantitative Structure–Activity Relationships (QSAR)] in an effort to find rapid, computational tools for developing molecules with particular biological activities. 150 As these tools were developed, it became obvious that molecular simulation in itself was insufficient to design new targets, but that it needed to be part of a larger, integrated effort that included data management and optimisation schemes. Quantitative structure–property relationship techniques have also been proposed for assisting in the evaluation and classification of potentially hazardous chemicals.62,151 A similar trend has arisen for the application of quantum mechanical techniques to designing inhibitor molecules. 47 The early methods parallelled the QSPR approach used in drug design – correlating inhibitor efficiencies with molecular descriptors that could be readily computed using quantum chemistry codes, such as the energy levels of the highest-occupied and lowest-unoccupied molecular orbitals (E HOMO and E LUMO).152–158 Similar QSPR approaches have been applied to the ranking of oxyanion inhibitors (i.e. molybdate, chromate, tungstate, nitrate, etc.). 159 Although not integrated into a mechanistic-based model, the approach was used to demonstrate that molecular descriptors show rough correlations with the general sequence of inhibitor efficiencies.
Despite the intensity of effort to develop linear and nonlinear expressions for corrosion inhibitor efficiency from the molecular descriptors – including an expanded set of descriptors that now includes Fukui indices that categorise individual atoms according to hard/soft acid/base theory 160 – it is not clear that the effort has led to breakthroughs in ultrahigh efficiency inhibitors. The power of QSPR approaches derives from the quantum mechanics adage that ‘everything is in the wavefunction’, however, reducing the wavefunction to a finite set of descriptors does not guarantee that inhibitor efficiency will emerge from a regression. More recent approaches to the molecular modelling of inhibitor species, therefore, are moving towards the modelling of the mechanism of inhibition, and include factors such as the binding energy of the inhibitor to model metal or metal-oxy-hydroxide surfaces.48,161–168 An early molecular approach that would be worth revisiting in this regard is the simulation of packing factors based on molecular geometry considerations. 169 The trend represents a step towards the integrated approach that bridges quantum mechanical methods with density functional theory and atomistic simulation and high-level surface characterisation, with the next step being a bridge to electrochemical models for the inhibition of anodic and/or cathodic reactions. Additional features that should be considered in an integrated approach include the combination of inhibitors with other surfactants and additives, intrinsic inhibitor qualities of crudes, partitioning between oil and liquid phases, the effect of inhibitor molecules on multiphase flow, and the evolution of inhibitor efficiency over time.170–172 Once the ability to compute inhibitor efficiencies based on molecular structure and a given material/environment combination develops, then the computer-aided design of optimum inhibitor molecules will be possible, according to the general optimisation/calculation approach outlined in Fig. 4 and introduced in further detail below.
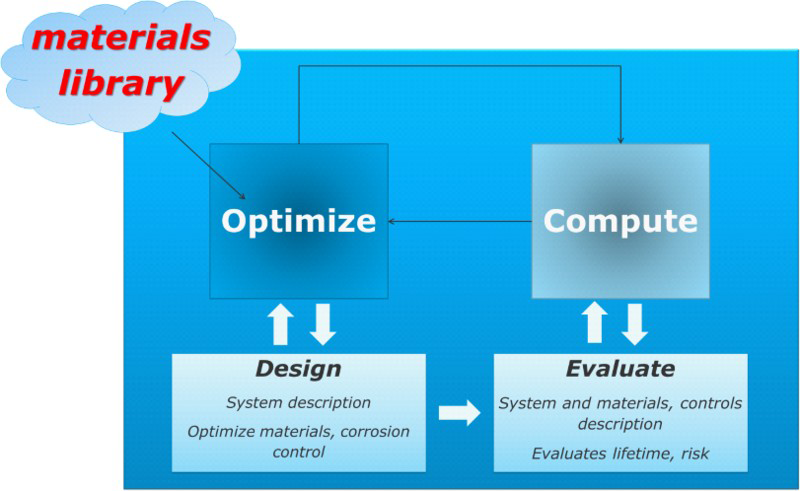
Accelerated discovery requires the integration of an optimisation loop with an embedded computation scheme, with access to a library of materials features that can be controlled. In the case of accelerated design of corrosion-resistant systems, the optimisation loop may vary materials parameters and corrosion control solutions (coatings and inhibitors, for example) in order to minimise the risks associated with failure by corrosion, or to maximise the usable lifetime
At the macroscopic length scale, finite element modelling was applied to simulate the electrochemical environment surrounding subsea pipelines. 173 The breadth of the model allows it to take into account the geometry of the pipeline, and the effects of sea depth, anodic bracelets and defects (i.e. areas in which the coating was breached). The method was used to predict variations in potential and corrosion current along the line, and then comparison was made with inspection data. Peaks in the inspection data correlated with expected areas of uncoated surface, in this example, the peaks corresponded to the position of flanges.
Electrochemical simulations have been used to model the localised corrosion of mild steel occurring at a holiday in a zinc-based coating. 174 The one-dimensional electrochemical simulation was evaluated numerically to predict the time required for the pit to extend to 1 μm using a computationally expedient algorithm. This simulation was then embedded into a large-scale optimisation scheme, which targeted the development of an optimal coating composition graded across the depth of the coating. Different coating treatments were specified by their shift in the anodic potential relative to pure zinc, and gradings were simulated by changing the variation of this parameter across the coating. A simulated annealing procedure was then used to optimise the coating parameters based on the aforementioned resistance criterion. The model was used by the authors to present an optimal grading for the coating; however, experimental validation was not performed to test the predictions of the model.
This kind of model can be generalised as consisting of a computation scheme (in this case, for a metric that indicates the degree of corrosion – the time to pit to 1 μm) embedded inside an optimisation scheme (to produce an optimal coating), see Fig. 4. To achieve the goals of the materials genome initiative – i.e. reduction of the time and costs associated with materials development – it is therefore necessary to address the three elements of (a) the computation scheme (in this case, the method for computing the corrosion performance of the system), (b) the optimisation scheme (in this case, a simulated annealing approach for varying the coating parameters), and (c) the library of materials parameters, which are available for optimisation (here, the anodic potential of the coating). For practical corrosion lifetime prediction of existing systems, it may be sufficient to focus on the computation scheme, and, as reviewed in this work, there remains significant progress to be made in this area. However, when looking towards design and the deployment of systems into new environments, it will be necessary to embed high-fidelity corrosion models into an optimisation module. In this way, materials parameters – such as alloy composition and surface treatment – could be optimised along with other features such as environmental controls, and the application of coatings and inhibitors. From a fundamental perspective, optimisation is the inverse problem to property computation, and, outside of indirect, extensive search methods, much more challenging to directly solve. 175
Deterministic models for probabilistic failures
Although the processes leading to corrosion follow a series of mechanisms that can be, in theory, simulated according to the laws of physics, corrosion phenomena are highly variable and this observation has led to a preference in some groups for the application of probabilistic models for corrosion prediction. The idea that uncertainty can arise from otherwise deterministic processes is not unusual: sources of uncertainty arise from the incomplete knowledge regarding boundary conditions, the interactions between processes that may have been assumed to be independent, the omission of details from a fundamental model, or the variations in the material and environment parameters themselves: microstructure, turbulence, compositional variations, and third-party damage (Fig. 5). Thus, despite the best of efforts to yield a numerical or analytical prediction, there will always arise some uncertainty because of the distribution of conditions that exist in the real world, and the inability for any given model to include all the possible physical processes that could be occurring in sequence and parallel.

Schematic illustrating how uncertainties in the input variables, x 1 and x 2, to a deterministic model have consequences upon the model prediction, y. Furthermore, additional uncertainty in y is introduced from the assumptions and incompleteness of the model itself, although this second kind of uncertainty is usually more difficult to assess
A recent example of the use of statistics to capture the variation in corrosion performance concerns the corrosion wastage of ballast tanks by sea water. 29 A Weibull distribution was used to describe the statistical distribution of corrosion depth for each of the time intervals surveyed. A polynomial equation was then fitted to the Weibull distribution parameters α(t) and β(t) to provide a Weibull distribution function that varied with time. The advantage of such an empirical approach is that it was able to utilise real field data and directly incorporated probability distributions that quantified the uncertainty and allowed for the extreme statistics of the corrosion events. On the other hand, such a purely empirical approach leads to fits that have dubious reliability, such that extrapolation beyond the time-series used to fit the data should be done with extreme caution. Furthermore, the lack of physical understanding means that when unknown conditions of environment or alloy are encountered, the model becomes irrelevant. The advantage of incorporating a physics-based interpretation to the predictive model is that the laws of physics are time-invariant, and thus provide a sounder basis for extrapolation.
The challenge to reconciling statistical and deterministic models for corrosion was directly recognised by Engelhardt and Macdonald, who developed a damage function approach that attempted to combine the two. 28 A damage function analysis was implemented for crack pit propagation based on the diffusion flux. Simplifications were made to allow for an easy derivation, but the more complex forms that would account for variable nucleation, propagation and repassivation rates were also provided, as well as statistical variations in the model parameters using a Laplace distribution. It was shown that, in the limiting case, the damage function analysis provides analogous expressions to the Gumbel type 1 extreme value statistics. The point defect model of Macdonald was used to provide some analytical forms for the nucleation of metastable pits, and it was assumed that a proportion of such pits would become active.
A third approach to incorporating probabilistic effects into models comes from reliability theory and the use of first- and second-order reliability methods to explore the consequences uncertainty in the input parameters has on the model outputs. This approach was used to incorporate an uncertainty analysis into a deterministic model for chloride-induced corrosion under concrete. The model predicted the extent of corrosion given the parameters concrete cover depth, chloride diffusion coefficient, chloride threshold value, and surface chloride concentration. 17 The authors recognised two sources of uncertainty: (a) uncertainty in the parameters (aleatoric uncertainty, associated with the intrinsic randomness in the data) and (b) epistemic uncertainties that derived from a recognition that any physics-based model will be incomplete as regarding all of the factors influencing the mechanism of attack. Uncertainty can also be introduced through the process of measurement, and the choice of the probabilistic submodel (that is, the probability distribution used to model the basic variables). The authors sought to treat uncertainty using three methods: first- and second-order reliability methods that expand the probability distribution in terms of a first- or second-order Taylor function, and Monte Carlo simulation. The authors showed that the reliability methods provided an equivalent evaluation of the uncertainty and were able to produce an answer much faster than performing a large number of Monte Carlo simulations. The first-order reliability method only required the mean and standard deviation of the input variables, whereas the Monte Carlo methods required the generation of an entire probability distribution. Hence, it appeared from this work that the first-order reliability method could provide a fast way to ascribe uncertainty to the predictions of deterministic models, given some expected variations in the input parameters.
A similar effort to reconcile mechanistic models dependent on distributed variables was made by Harlow and Wei. 176 The authors considered corrosion and fatigue response of aluminium and steel alloys by developing damage accumulation functions for pitting and fatigue cracking. The damage accumulation function approach used a representative function to track the changes in microstructural properties with loading and environment as a means for quantifying the progress made towards materials failure. The functions themselves incorporated both mechanistic and probabilistic aspects. The authors used the Monte Carlo method to sample the variation of the model input parameters, while acknowledging that the method inevitably performs poorly for tail regions. A pitting model was developed for the distribution of inclusions in the material, and a hemispherical pitting growth model was applied (i.e. a deterministic, electrochemical model for pit growth). The probability distribution functions were introduced for the random variables: pit currents, the number of particles and particle size. The corrosion fatigue crack growth rate was treated by a power law. The criterion controlling pit-to-crack transitions was built through the mechanistic model that related pit size to the crack growth rate. The authors then used this model to predict the critical size and the time to failure. This simple, binary model (i.e. two key mechanisms contributing to failure) has one to two orders of magnitude of random variables, thus creating already a complicated probabilistic formulation that could only be solved through numerical simulation. The random variables controlling failure and incorporated into the model were directly assessed by materials characterisation and found to be a function of the processing conditions. As a result, a probability distribution was constructed for the particle cluster size. Weibull distributions were used for many of the variables based on previous experimental analyses. The model was then used to investigate the failure, and it was found to work well for relatively small cracks, but less so far larger cracks. Such methods demonstrate the power of coupling probabilistic models with the deterministic, but required significant effort to accurately estimate the statistical characteristics of the material properties.
As mentioned above, in addition to uncertainty that arises from variation in a model input parameter, uncertainty also derives from the knowledge limitations of the model itself. Although it is difficult to quantify the uncertainties that arise from ‘unknown unknowns’ – i.e. the mechanisms one has not included or does not know how to include – uncertainties can be introduced because of the approximations in numerical solvers. Model uncertainties of this nature occur at all levels in the modelling hierarchy. For example, Grossfield and Zuckerman reviewed the uncertainties that arise from molecular dynamic and Monte Carlo simulations of biomolecules: techniques that sample the active space of a molecule, but because of the requirements that simulation time be finite, the extent to which a simulation can be considered adequately sampled needs to be quantified.
82
Most simulations are confined to short timelines (pico- to micro-seconds of real molecular time, at best). The authors derived some ‘rules of thumb’ for assessing the statistical quality of such simulations:
Perform multiple, independent simulations. Use block averaging to estimate uncertainty. Principle component analysis (PCA) or all-to-all root-mean-square deviation (RMSD) plots will indicate the number of independent states assessed in the simulation. Investigate structural decorrelation to estimate the slowest timescale affecting simulations, and for non-dynamics, use variance analysis. Consider a smaller, better sampled model instead of a large, poorly sampled model system.
A more rigorous formulation was outlined by Strachan et al. and applied to the physics-based simulation of materials deformation phenomena via finite element methods.
20
By investigating parameter sensitivities to the level of physical rigour applied in such simulations, the most optimal points for performing high-fidelity simulations were decided (using a chain-law approach with functional derivatives) such that a correction could be made to the ‘quantities of interest’ calculated from low-fidelity simulations. The technique of functional derivatives was used to bring an element of rigour and quantification to the epistemic error associated with using ‘coarse-grained’ physical simulation or analytical techniques for efficient, but approximate, problem solving. Similarly, in quantum mechanical calculations, methods have been developed for probing the magnitude of errors associated with fundamental parameters, such as the number of basis set functions used to construct the molecular wavefunction.177–183
An ambitious model for integrating probabilistic and deterministic approaches to corrosion simulation utilised the method of Bayesian networks.65–67 By following corrosion models for pipeline systems available in the literature, a network was created that connects the probabilities for numerous system variables to provide an overall prediction of corrosion risk, as a function of position and time, with quantified uncertainty. Corrosion mechanisms included were internal and external corrosion, high and near-neutral pH SCC and the influence of cathodic protection and surface coatings. Other diverse phenomena that the method included using the probabilistic treatment were the placement of welds, the materials properties such as surface treatment and coating quality, residual stresses, seasonality, and soil types. The advantage of such an interconnected model based on Bayesian inference was that expert opinion (or even estimated parameters) could be reconciled with deterministic models in one overall framework. Inference techniques can also be used to identify the leading sources of error in the interpretation of experimental measurements and to develop practices for optimal experimental design. 184
The future of corrosion informatics
A review of this nature can only scratch the surface of the numerous modelling efforts taking place in the field of corrosion science and engineering, and, for this reason, many significant models will have been overlooked. This incompleteness in itself is a contributor to the challenge facing the materials community. The information era has enabled data generation of an unprecedented quantity and rate, and efforts across research groups globally continue to produce models that aim to make sense of the data, in turn, generating more data that need to be qualified and assessed. Corrosion informatics, the purposeful integration, organisation and analysis of both data and models provide the opportunity to obtain significant gains in the quality of lifetime predictions and the principles for materials and system design, but the challenge is how.
When considering the development of integrated, high-fidelity lifetime prediction models for corrosion and related phenomena, careful thought needs to be given to the appropriate degree required for each element of the problem. For instance, given the task of predicting corrosion in a pipeline that spans hundreds of kilometres, would an atomistic representation be useful, let alone tractable (Fig. 6) 185 Considering that 1 km of pipeline could include approximately 1015 μm-sized grains and precipitates, with a similar order of magnitude of grain boundaries, and each grain comprising ∼1015 atoms, even a simulation that operates at the mesoscopic level of grains would be too complex to be computationally tractable. Instead, it makes more sense to decompose the corrosion phenomena into distinct length scales according to their heterogeneity. For instance, megascale conditions, such as local climates might vary on the length scale of 10s of kilometres. Soil quality, on the other hand, might vary on the order of metres. Galvanic cells and other electrochemical conditions would vary on the level of centimetres to millimetres. Microstructural variations within the material occur on the length of the grain size, sub-micrometres to tens of micrometres. Atomic scale heterogeneities, including point-, line- and planar-defects take place over nanometre and sub-nanometre scales. Thus, it would suffice to explore, using advanced characterisation and ab initio models, the contributors of microstructural effects under the possible set of variations that may be introduced at the higher length scales. Since the materials microstructure is likely to remain similar at points along the multi-kilometre stretches of material, appropriate parameters from the lower length scale could be used in the higher length scale models. Thus, the hierarchical approach to multiscale modelling provides an efficient pathway to obtain an integrated approach. A similar breakdown and integrated multiscale approach has been proposed by Cole and Marney, using the four subcategories of electrochemical activity, oxide scales, local macro-environmental parameters (e.g. soil pH), and general external environmental factors (e.g. seasonal variations), drawing from the extensive models developed by soil scientists and hydrologists. 186
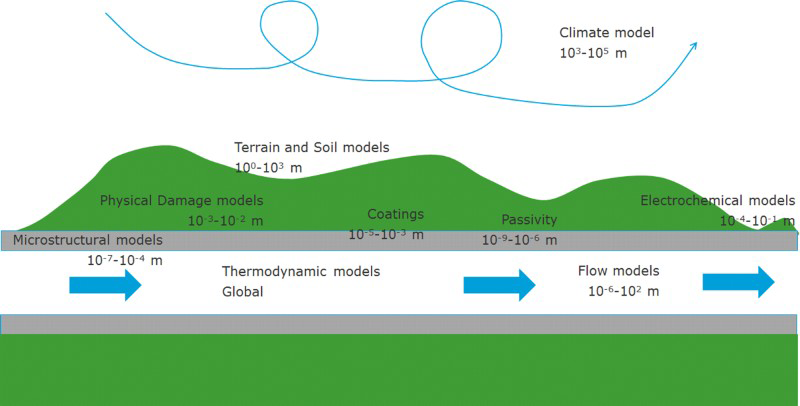
Illustration of the integrated modelling approach to pipeline corrosion, showing a number of contributing factors that are amenable to modelling at various length scales
This approach follows the principle of domains and microprocesses described by Staehle. 128 It should be accompanied by an uncertainty analysis, whereby the models from each domain are scrutinised for those sources of uncertainty that arise because of model parameterisation, approximations contained within the model, and for those physicochemical and metallurgical features that are not addressed, or only addressed with partial knowledge. 21 In this way, those aspects that the model can confidently address are separated from those aspects that are partially or incompletely addressed, and the domains and microprocesses that are ignored or conjectured to occur but for which models are not yet available are also specified. With this last point in mind, it could be said that it is just as important to confess to what is unknown as it is to describe what is known. 187 This point is particularly critical when linking models together, as uncertainties at lower length and timescales can propagate upwards to become much more significant sources of error when they are inserted into higher length and/or timescale codes.
As modelling, experiment and sensor technologies continue to inform corrosion science and engineering, the informatics approach will become more and more necessary. This review outlines several different subcategories with a focus on modelling technologies and outlines particular areas that are in need of further development. To summarise these areas include:
Categorisation of models into domains and microprocesses and establish the linkages between them via multiphysics integration. Improvements in this area will come about through interdisciplinary collaborations between the sciences and interactions between characterisation and modelling groups. Better quantification of the uncertainties arising from all sources of information, including the application of models, raw data from the field and experimental testing. More incorporation of statistical approaches, including Bayesian analysis and reliability methods with both physics-based and empirical modelling will advance this point. Promotion of data and model sharing to the extent possible while maintaining the bounds of intellectual property. This could include data sharing within organisations and/or independent third parties. Engagement with effort such as the materials genome initiative will help in the development of standard data formats and libraries to collate relevant models and fundamental parameters.
188
Footnotes
Acknowledgements
Support for this project is acknowledged from the Strategic Research and Innovation group within DNV GL. The author thanks Narasi Sridhar (DNV GL) for insightful discussions and a careful reading of the manuscript.