
Abstract
The effect of chloride ions’ presence (0·005-1·0M NaCl) in phosphoric acid solutions (5, 40 and 75%) on the corrosion behaviour of three austenitic stainless steels (an experimental steel Fe–18Cr–12Mn–0·6N and two trade grades, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N) has been studied by potentiodynamic polarisation measurements. The surface examinations of the samples tested involved X-ray photoelectron spectroscopy as well as optical and scanning electron microscopy. It was established that chlorides added to phosphoric acid solutions deteriorate the general corrosion resistance, and under anodic polarisation, they provoke pitting corrosion. The composition of the stainless steels significantly influences its corrosion behaviour in the phosphoric acid solutions containing chloride ions. The replacement of nickel with manganese and nitrogen on top of lower chromium content has a strong negative effect on the corrosion resistance.
Introduction
Austenitic nickel free or of reduced nickel content stainless steels are under intensive investigations because of their low cost production and enhanced mechanical characteristics with respect to the conventional chromium–nickel counterparts. The reduction of nickel or its complete elimination in the austenitic stainless steels can be attained through its replacement by other austenite forming elements as nitrogen or/and manganese. Such steels are known as chromium–manganese austenitic stainless steels (grades 200 series), and they are encountered in many engineering applications owing to the great mechanical properties combined with acceptable corrosion resistance.1,2
Stainless steels demonstrate good corrosion resistance in neutral and alkaline environments because of spontaneous formation of passive protective layers. Nevertheless, such steels are vulnerable to a local corrosion when aggressive ions, for instance chlorides, destroying the passive layers exist in the environment. The latter is strongly expressed when stainless steels are placed in acid environments containing halogen since most of them are highly susceptible to pitting corrosion in these conditions. Phosphoric acid solutions are one of the cases of massively produced and widely used environments which could be contaminated with chlorides.
However, systematic studies on the corrosion resistance of iron and ferroalloys and in particular of chromium–manganese–nitrogen stainless steels in acid phosphate environments containing aggressive ions are rare in the scientific literature.3–9 Some of these works determine the effect of temperature and of various contaminants of the phosphoric acid such as Cl− , S2 − as well as other species in small quantities emerging in the production process.10–14 The acid concentration in these studies was around 40-50%.11,13
In contrast, there are many sources of information about the inhibition effect of phosphate ions on the pitting corrosion of steels in chloride environments. In most of these studies, the model environments are either neutral or alkaline chloride solutions with low phosphate ion contents.15–20 Browsing published results relevant to the effect of phosphates on the passive states of steels and consequently their pitting corrosion resistance, many contradictory outcomes can be encountered. According to some authors,15,16,21 the phosphate ions suppress the formation of pits to a greater extent than in cases of chromate, sulphate and nitrate ions. The explanations of such strong inhibition effect are not based on a unified concept. Refaey et al.,16,21 for example, suggest that the phosphate ions’ action is as a result of deposition of insoluble compounds on the steel surface, while Zuo et al.
15
explain the effect of the phosphate ions by a decrease in solution acidity inside the pits as a result of hydrolysis processes related to 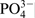 . The reduction of the local solution acidification decreases the rate of metastable pit nucleation and delays the pit growth owing to the repassivation processes. The same authors drew their conclusions from experiments in environments modelling inner pit solutions (4·13M NaCl and pH 1). The experiments revealed that, even for phosphate/chloride ratio of 1:250 in the environment, the tested stainless steel 316 remained in spontaneous passive state with increased width of the passive region. In addition, the passive current was lower than in cases when phosphates are missing. In contrast to these establishments, in similar studies on stainless steel 304 behaviour in environments containing phosphate ions of low concentration (0·014M Na2HPO4), it was reported by Lakatos-Varsányi et al.
19
that there were no effects of the phosphates on the current in either the active and the passive regions, and the steel pitting formation potential remained unaffected. The only reported positive effect by these authors was related to the repassivation potential, and the explanations were again based on the limitation of the pit solution acidity.
. The reduction of the local solution acidification decreases the rate of metastable pit nucleation and delays the pit growth owing to the repassivation processes. The same authors drew their conclusions from experiments in environments modelling inner pit solutions (4·13M NaCl and pH 1). The experiments revealed that, even for phosphate/chloride ratio of 1:250 in the environment, the tested stainless steel 316 remained in spontaneous passive state with increased width of the passive region. In addition, the passive current was lower than in cases when phosphates are missing. In contrast to these establishments, in similar studies on stainless steel 304 behaviour in environments containing phosphate ions of low concentration (0·014M Na2HPO4), it was reported by Lakatos-Varsányi et al.
19
that there were no effects of the phosphates on the current in either the active and the passive regions, and the steel pitting formation potential remained unaffected. The only reported positive effect by these authors was related to the repassivation potential, and the explanations were again based on the limitation of the pit solution acidity.
The present work takes into consideration the effect of the chloride ions’ presence in phosphoric acid solutions on the corrosion behaviour of three austenitic stainless steels: an experimental steel Fe–18Cr–12Mn–0·6N and two trade grades, i.e. AISI 304 and the Russian X14AΓ15. Precisely, the study focuses on the effect of experimental steel composition and its relationships to the exhibited corrosion resistance, compared to those of conventional nickel steel and manganese–nitrogen steel samples.
Experimental
Austenitic stainless steels Fe–18Cr–12Mn–0·6N, Fe–14Cr–15Mn–0·2N and Fe–18Cr–9Ni were used in the tests. Their chemical compositions are summarised in Table 1. All samples were thermomechanically treated under almost similar conditions to achieve austenitic structures. Oxides and oxysulphides were detected as principal non-metallic inclusions in the steel samples. The chromium–manganese–nitrogen steels are richer of sulphide inclusions than the chromium–nickel steel.
Chemical compositions of stainless steels/wt-%
Samples with a contact surface area of ∼0·5 cm2 were first wet ground with 120-600 grit silicon carbon papers. Before polarisation tests, the working electrode was ground with 800 abrasive paper, then degreased in alcohol–ether mixture and washed in distilled water. Before the potentiodynamic polarisation tests, the surfaces of the samples were polarised for 5 min at − 0·7 V(SCE) in order to remove any air formed oxides.
The test environments were phosphoric acid solutions of concentrations 5, 40 and 75%, with added amounts of 0·005-1·0M NaCl prepared by dilution with monodistilled water; the chemicals used were 85 wt-% phosphoric acid and sodium chloride of class p.a.
Open circuit potential (OCP) was measured during 1 h in the test solutions, and the values at the end of the period were averaged.
Cyclic potentiodynamic polarisation measurements were performed with a potential scan rate 1 mV s− 1, starting from the potential − 0·7 V(SCE) followed by anodic polarisation. After passing through the passive state and the current density exceeded one order the passive current density, a polarisation in the reverse (negative potential direction) was carried out down to the point where the curve intersected the anodic one. Every potentiodynamic polarisation test was performed five times. The results were arranged from lowest to highest value, and the middle one was considered as final. After accomplishment of the individual potentiodynamic tests, the sample surfaces were observed with an optical microscope at magnification × 200. In cases were crevices exist at the sample/isolation interfaces, the tests results were ignored.
Electrochemical polarisation curves were obtained with a galvanostat/potentiostat (Princeton Applied Research Model 263) and computer software Power Suite. The electrochemical measurements were carried out in a conventional three-electrode cell with a platinum counter electrode and a saturated calomel electrode (SCE) as a reference electrode. All potentials were reported with respect to the SCE. The experiments were carried out at room temperature in naturally aerated solution without stirring.
The film composition of Fe–18Cr–12Mn–0·6N after an anodic polarisation at 0·4 V(SCE) for 2 h in 40 wt-%H3PO4 (with and without addition of 0·1M NaCl) was investigated by X-ray photoelectron spectroscopy. The measurements were performed in a VG ESCALAB II system using Mg K α radiation with energy of 1253·6 eV. The pressure in the chamber was 10− 8 Pa. The binding energies were determined with an accuracy of ± 0·1 eV utilising the C 1s line at 285·0 eV (from an adventitious carbon) as a reference. The changes in composition and chemical surrounding in the depth of the films were determined on the basis of the areas and binding energies of C 1s, O 1s, N 1s, Fe 2p3/2, Cr 2p3/2, Mn 2p3/2, P 2p and Cr 2p3/2 photoelectron peaks (after subtracting the background using the Shirley) and Scofield's photoionisation cross-sections.
Results and discussion
Potentiodynamic polarisation study
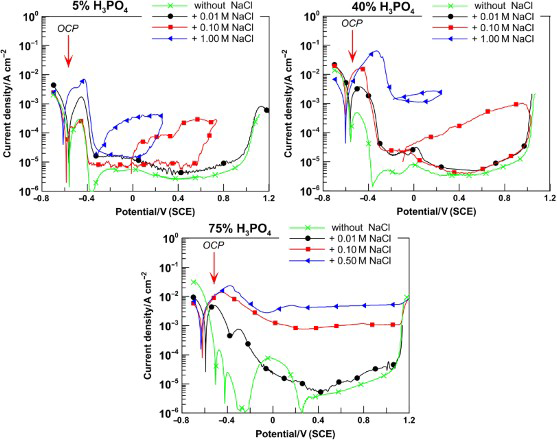
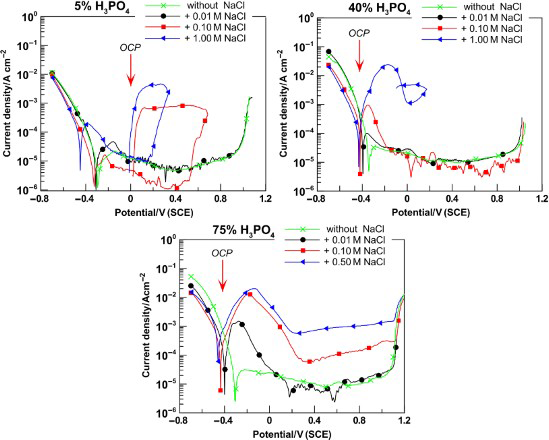
The effect of chloride ions on the corrosion behaviour of the steels in phosphoric acid was examined in three different acid concentrations: low, 5%; medium, 40%; and high, 75%. The typical polarisation curves are shown in Figs. 1 and 2 for Fe–18Cr–12Mn–0·6N and Fe–18Cr–9Ni steels respectively. For the tests with 75%H3PO4, the highest chloride concentration was 0·5M in order to avoid saturation of the solution. The polarisation relationships reveal the main electrochemical corrosion parameters providing information about:
active region and general corrosion behaviour (corrosion potential E corr and corrosion rate i corr) active–passive transition (critical potential E cr and critical current density i cr) passivity and passivity breakdown (passive current density i pass, pitting potential E pit and repassivation potential E rp). Polarisation curves of Fe–18Cr–12Mn–0·6N steel in solutions of 5, 40 and 75%H3PO4 containing NaCl; 1 mV s− 1, 20°C Polarisation curves of Fe–18Cr–9Ni steel in solutions of 5, 40 and 75%H3PO4 containing NaCl; 1 mV s− 1, 20°C
General corrosion behaviour
The investigation of stainless steels behaviour began with obtaining the OCP versus time dependencies. It was established that in all phosphoric acid solutions with the presence of sodium chloride, OCP moves in negative direction and stabilise in ∼5 min. This behaviour could be explained by the initially dissolving of air formed passive film followed by steel matrix dissolution. After 1 h, the samples’ surfaces were covered with dark, uneven porous layer from corrosion products. The latter indicates that steels remain in active state in this environment. There was only one exception for the samples of Fe–18Cr–9Ni steel in 5%H3PO4, containing chloride up to 0·1M, for which there was ennoblement of potential and clear surface after 1 h of immersion. The stationary values of OCP after 1 h stay in phosphoric acid solutions with 0·1M NaCl are mentioned on the polarisation dependencies (Figs. 1 and 2). It is obvious that OCP values are ∼50 mV more positive than the potentiodynamically measured corrosion potentials and fall within the area of active dissolution.
As a strong attack, and therefore irreproducible changes, in the steel surfaces at open circuit conditions takes place, polarisation dependencies were taken without prior stay in corrosion environment. Only short cathodic treatment was carried out, during which residues of air formed passive layers were removed without dissolution of metal surface. From as received polarisation dependencies, it was established that, in all phosphoric acid solutions containing chloride ions, all stainless steel samples tested are in active state. This can be judged from the negative values of the corrosion potential and clear active–passive transition.
The corrosion potential evolution affected by chloride ions added to the phosphoric acid solutions is presented in Fig. 3. The corrosion potentials of all samples shift in the negative direction, in accordance with the equation 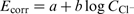 . The b slopes of all curves are about 20-30 mV/decade. As an exception, it can be seen that the corrosion potentials E corr determined in solutions of 5%H3PO4 with chloride content up to 0·1M have relatively constant values. Similar shifts of E corr in the negative direction with increase in chloride concentration were reported for steel 316 in 30%H3PO4.
6
Moreover, in the case of OCP and 30%H3PO4 with 1·5%NaCl, the test with steel 316 revealed predominant dissolutions of Cr and Fe components with simultaneous enrichment of the steel surface with nickel.
7
Under these conditions, the passive layer thicknesses were smaller than those formed in the absence of chlorides in the acid solutions.
. The b slopes of all curves are about 20-30 mV/decade. As an exception, it can be seen that the corrosion potentials E corr determined in solutions of 5%H3PO4 with chloride content up to 0·1M have relatively constant values. Similar shifts of E corr in the negative direction with increase in chloride concentration were reported for steel 316 in 30%H3PO4.
6
Moreover, in the case of OCP and 30%H3PO4 with 1·5%NaCl, the test with steel 316 revealed predominant dissolutions of Cr and Fe components with simultaneous enrichment of the steel surface with nickel.
7
Under these conditions, the passive layer thicknesses were smaller than those formed in the absence of chlorides in the acid solutions.
Corrosion potential as function of NaCl concentration in 5, 40 and 75%H3PO4 for Fe–18Cr–12Mn–0·6N, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N steels
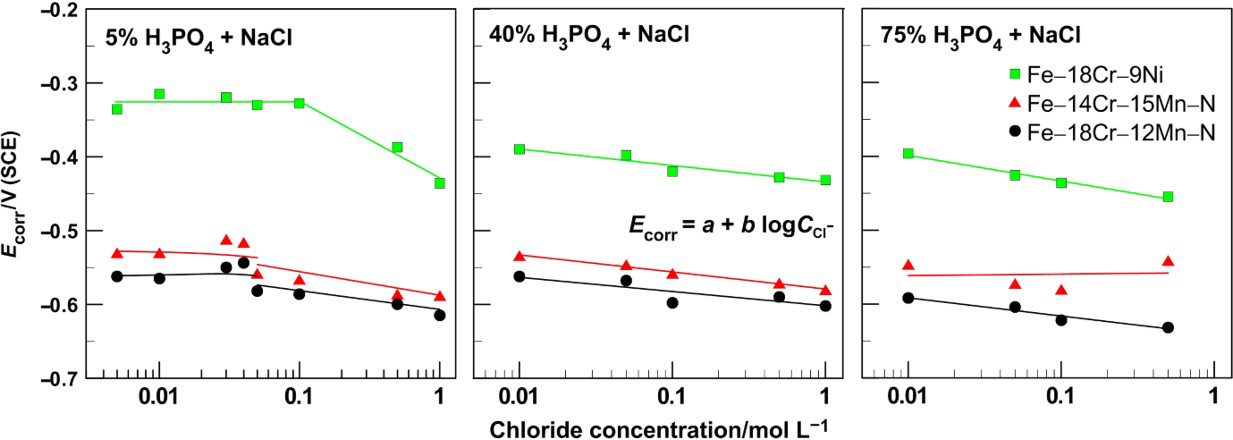
The corrosion rates (expressed by the corrosion current density) as functions of the NaCl concentration in 5, 40 and 75% phosphoric acid solutions are shown in Fig. 4. The increase in the chloride ion concentration accelerates the general corrosion rate in acid solutions, and this effect is manifested markedly when the NaCl concentration exceeds 0·1M in 5% acid. Precisely, in a solution of 5%H3PO4 with added 1·0M NaCl, the corrosion current for samples of Fe–18Cr–12Mn–0·6N steel reaches its maximum of ∼8·4 Ma cm− 2. This maximal corrosion current is about one order higher than that observed at low chloride concentrations and about two orders higher than that recorded in 5%H3PO4 in the absence of chlorides. 22 Further, in 40 and 75% phosphoric acid solutions, the addition of chlorides accelerates the corrosion rate, and the corrosion current densities increase about two to three times within the studied concentration range.
Corrosion current density as function of NaCl concentration in 5, 40 and 75%H3PO4 for Fe–18Cr–12Mn–0·6N, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N steels
Similar negative effect of the chloride ions on the i corr is registered also by Refaey 21 for steel corrosion in hydrochloric acid solutions. Refaey explains the results by a synergetic effect between Cl− ions chemisorbed on the metal surface and H+ ions from the solution, which are electrostatically attracted by Cl− . The result is a layer of ions adsorbed to the surface acting as a catalyst for the steel dissolution reaction. 21
Active–passive transition
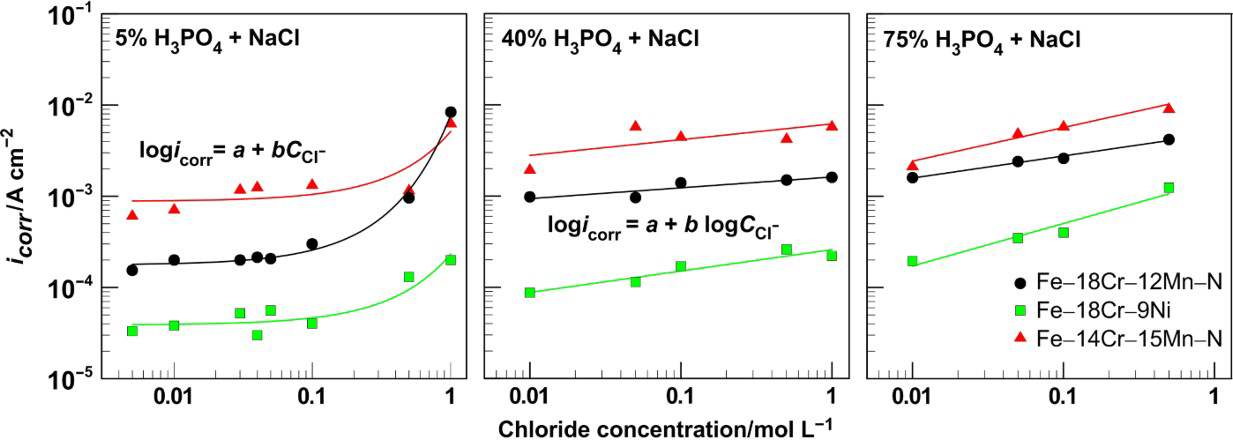
The addition of chloride ions into phosphoric acid solutions hampers the transition to anodic passivity state for all steel samples tested. In general, the critical current density (Fig. 5) as well as the distinction [E cr − E corr] (Fig. 6) grows up. The values of i cr and E cr obtained with 5%H3PO4 scatter (see Figs. 5a and 6a), while with increase in the phosphoric acid concentration, the points depend almost linearly on the concentration (Figs. 5b and 6c) or slightly deviate from linear relationships (Figs. 5c and 6b). Similar rapid increase in i cr and a shift of E cr in the positive direction with increase in NaCl concentration have been reported for steel 316 in 30%H3PO4. 6
Critical current density as function of NaCl concentration in 5, 40 and 75%H3PO4 for Fe–18Cr–12Mn–0·6N, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N steels
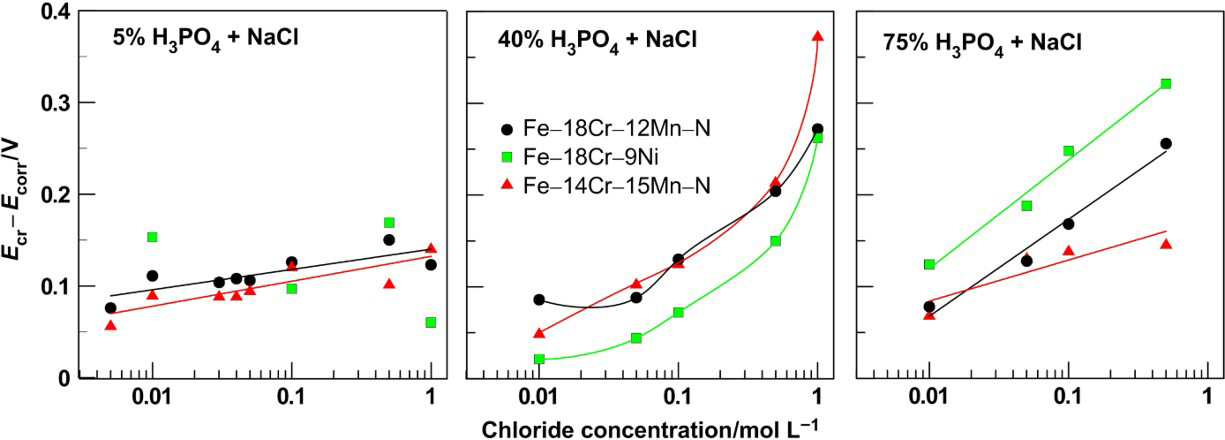
Difference between critical and corrosion potentials as function of NaCl concentration for Fe–18Cr–12Mn–0·6N, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N steels in 5, 40 and 75%H3PO4
It is interesting to note the fact that the presence of chlorides in the phosphoric acid affects most unfavourably the anode behaviour of the chromium–nickel steel. In solutions not containing chloride ions when anode polarisation is applied, the chromium–nickel steel is in state of spontaneous passivity (Fig. 2). At the same time, the chromium–manganese–nitrogen steels pass through active–passive transition (Fig. 1). In the presence of chloride ions in the solutions, the samples of Fe–18Cr–9Ni steel also begin to pass through active–passive transition when anodic polarisation is applied. Moreover, when the acid concentration is increased, then a well expressed peak of the anodic current for this steel is observed at lower chloride concentrations.
The critical current densities for Fe–18Cr–12Mn–0·6N steel samples are a little bit lower that those of Fe–14Cr–15Mn–0·2N steel but remain still higher with respect to the values established for the chromium–nickel steel. The difference in the values of i cr for chromium–manganese–nitrogen steels and chromium–nickel steel is well manifested in solutions with low chloride concentrations. In 75% acid with chloride content beyond 0·1M, the samples of the three steels exhibit almost equal i cr values.
Curiously, in pure phosphoric acid for all studied concentrations on the polarisation dependencies of chromium–manganese–nitrogen steels only, a second corrosion peak, at around − 0·4 V(SCE), was registered at the end of the active–passive area. It can be considered as a result of cathodic reaction of hydrogen evolution onto the passive surface. 23 The registered corrosion current is low and commensurable with passive current. Because of the high hydrogen ion concentration and the high environment viscosity, the participation of oxygen in this naturally aerated system is negligible. As opposed to the other authors, 23 the results received by us reveal that the chloride addition even in minimal quantity leads to formation of stable passive system and complete predominance of the anodic reaction on the cathodic one.
Passivity, passivity breakdown and repassivation
The polarisation dependencies on Figs. 1 and 2 demonstrate typical for chromium austenitic steels’ anodic passivity. When the chloride content in phosphoric acid solutions is low (up to 0·1M), the passive current density attaints minimum value at potential 0·4 V(SCE). In addition, anodic polarisation oxidation of Fe2+ to Fe3+ occurs, which results in current density increase. The state of anodic passivity of the studied steels starts at about − 0·2 V(SCE) and stretches to ∼1·0 V(SCE), where the transpassive dissolution and oxygen liberation begin. This stable passive state was observed for all three stainless steels tested (irrespective of the acid concentrations and chlorides content) below the critical value for pitting development (Figs. 1 and 2).
In all 5%H3PO4 solutions as well as in 40%H3PO4 with chloride content up to 0·1M, the passive current for Fe–18Cr–12Mn–0·6N and Fe–18Cr–9Ni steels holds in relatively stationary value of ∼10 μA cm− 2 (Fig. 7). The Fe–14Cr–15Mn–0·2N steel has a little bit higher passive current. In 40%H3PO4 with higher concentrations of active ions, the passive film protective properties considerably deteriorate. This behaviour is manifested by a sharp increase (more than two orders of magnitude) in the i pass value. Similar result has been reported by El Dahan 6 regarding 316 steel in 30%H3PO4: a sharp increase in the passive current at a concentration of 15 000 ppm NaCl (or 1·5% = 0·26M NaCl).
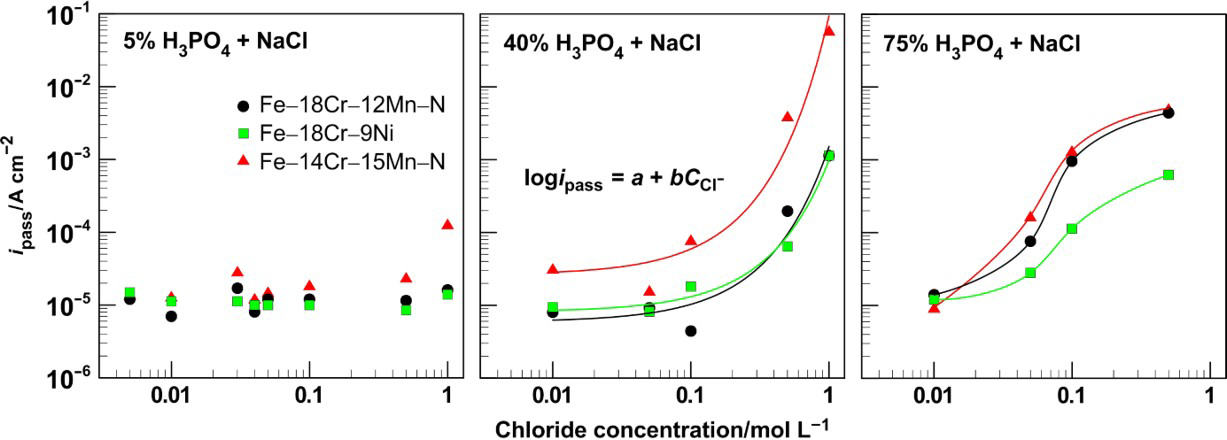
Passive current density as function of NaCl concentration in 5, 40 and 75% H3PO4 for Fe–18Cr–12Mn–0·6N, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N steels
The most strongly demonstrated negative effect of the chlorides was observed in the solutions of 75%H3PO4, where the passive current density i pass grew noticeably when 0·05M of NaCl was added to the solution. At higher acid and chloride concentrations, the passive current density reached ∼4 and 1 mA cm− 2 for chromium–manganese–nitrogen steels and the chromium–nickel one respectively without observation of pits formation. These high values of current density in the potentials corresponding to limiting current plateau indicate mass transfer controlled mechanism, in which electrochemical polishing occurs. The latter relates to high viscosity of 75% phosphoric acid combined with extreme aggression of the environment in respect to the passive layers on the steel surfaces.
In cases when the critical chloride concentration exceeds the threshold for pits formation, the passive region considerably shrinks as a result of the passive film breakdown at lower degree of anodic polarisation. The dependencies E pit–C NaCl for the three examined steels are shown in Fig. 8 and could be described in logarithmic coordinates, namely
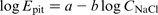
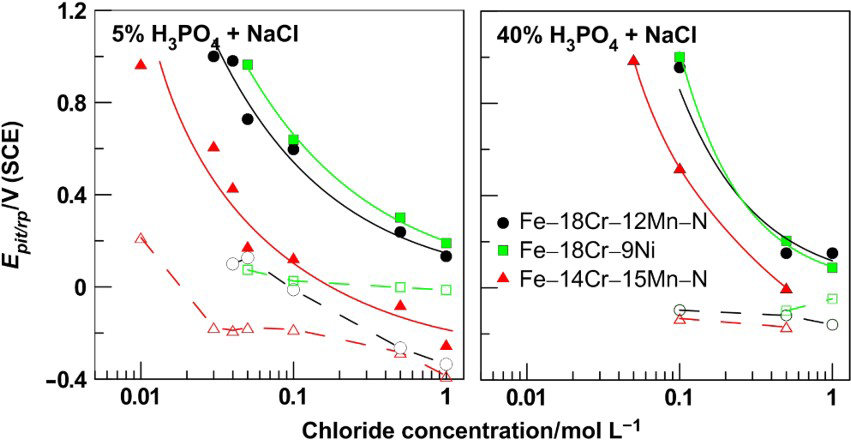
Passive layer breakdown potential (thick symbols and lines) and repassivation potential (dotted line) as function of NaCl concentration in 5, 40 and 75%H3PO4 for Fe–18Cr–12Mn–0·6N, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N steels
In 5%H3PO4 solutions, the pitting corrosion on Fe–18Cr–12Mn–0·6N, Fe–18Cr–9Ni and Fe–14Cr–15Mn–0·2N steels was developed when the chloride content was equal or exceeded 0·04, 0·05 and 0·01M respectively. The increase in acid concentration to 40% leads to a growth in critical chloride concentration. The stable pits development on the chromium–manganese–nitrogen steels in 40%H3PO4 was registered for concentration of 0·1M NaCl, but this was not enough to develop local corrosion damages when the chromium–nickel samples were tested in the same solutions. These results reveal that, with increase in the acid concentration, both the pit nucleation and growth were suppressed. The phosphate ions, however, despite the high aggressive environment with respect to the passive layers, inhibit the local corrosion activity.
The ability of the passive layer to recover itself from local breakdowns when the anodic polarisation reduces is related to the repassivation potential E rp, determined from the cross-point of the forward and the backward branches of the polarisation curve (Figs. 1 and 2). In general, the more positive the value of E rp, the easiest the return of the steel to the passive state. For the samples tested in this study, the repassivation ability of the passive layers in 5 and 40%H3PO4 deteriorates with the increase in the chloride concentration (Fig. 8); the effect is stronger in solutions of 5%H3PO4.
The results of potentiodynamic studies reveal that the steel's chemical composition significantly affects the corrosion resistance. In this context, the chromium–manganese–nitrogen samples demonstrate lower corrosion resistance than that exhibited by Fe–18Cr–9Ni steel samples in all tests carried out. The replacement of nickel by manganese leads to 150-200 mV more negative values of E corr and additionally to higher corrosion, critical and passive current densities. The lower chromium and nitrogen contents in Fe–14Cr–15Mn–0·2N steel samples as well as the increase in manganese content (compared to Fe–18Cr–12Mn–0·6N counterpart) also deteriorates the corrosion resistance.
These results could be taken as proof of some known results in the literature about the influence of manganese in other corrosion media. The added γ stabiliser manganese deteriorates the general corrosion resistance of different ferroalloys in non-oxidising acid environments, but does not have an effect on the passive current and the width of the passive region.22–26 However, the latter is true only for environments in which local corrosion does not take place. It is well known that the manganese non-metallic inclusions are anodes in respect to the steel matrix. Thus, in the presence of active ions, the increase in manganese content above 1% results in facilitation of pit formation processes and hampers repassivation ones. 27 Still, taking into consideration the low solubility of manganese phosphates, it was interesting to check the corrosion behaviour of high manganese stainless steels regarding its pitting corrosion resistance. One would expect that the manganese corrosion products could impede the pits’ growth in acid phosphate solutions, although at low pH value. Unfortunately, the received results refute these expectations. The steel with the highest manganese content, Fe–14Cr–15Mn–0·2N, is the most susceptible to pitting corrosion in 5 and 40%H3PO4 solutions. For the above mentioned steel, the most negative values of E pit and E rp were registered, as well as pit development at lowest chloride concentration compared with other investigated steels. The narrowness of the passive region is more likely to be because of lower Cr and N content than of the higher content of Mn. It is known that the nitrogen improves the general corrosion resistance of stainless steels in chloride environments acidified by sulphuric acid, but it is almost inactive when the environment is of neutral and alkaline nature.28–30 Moreover, nitrogen is shown to stabilise the passive state by facilitating the achievement of the passive state, decreasing the passive current and increasing the pitting corrosion resistance.29,31–33
Thus, it can be established that the steel with higher nitrogen and chromium contents exhibits better corrosion resistance than the other manganese steel, but lower than the nickel steel. The lower corrosion resistance of Fe–18Cr–12Mn–0·6N steel compared to that demonstrated by the chromium–nickel samples indicates that, despite a positive effect of its higher nitrogen content (0·61 wt-%), it is not enough to compensate the presence of manganese and the absence of nickel in the composition.
Microscopic study after electrochemical corrosion tests
The addition of chlorides above a concentration of 0·04M to 5%H3PO4 leads to local corrosion attacks of the surface, for all the three steels tested. With samples of Fe–14Cr–15Mn–0·2N, for instance, local breakdowns of the passive layer were observed at chloride concentration higher than 0·005M, although pitting corrosion development was not registered by the polarisation method. The most probable explanation for this behaviour is related to defects owing to dissolution of anodic non-metallic inclusions. 34 Similar defects in the passive films, to smaller extent, were observed with Fe–18Cr–12Mn–0·6N and Fe–18Cr–9Ni steels.
With increase in the chloride content in the solution, the number of pits on Fe–18Cr–12Mn–0·6N steel surface increases. The pit sizes decrease, and a general corrosion attack to the entire surface not affected by the pitting process starts. In 5%H3PO4 with chloride content of 0·05M, for instance, the number of pits observed was ∼10 cm− 2, of ∼120 μm in diameter, while the surrounding surface remained passive and shining. In addition, the test with 1·0M NaCl revealed an increase in the number of pits to several dozens: among them, micropits of dimensions below 30 μm predominated and were located mainly along the grain boundaries. Single open type large pits with deep irregular bottoms and mouths of ∼80 μm were also observed. Besides the pit formation, the corrosion attack affected the entire surface of the sample, which led to partial microetching of the structure. This effect probably occurs in the region of active dissolution where the kinetic controlled mechanism dominates. Additionally, the partial microetching of the structure was observed on the samples’ surfaces after OCP examination. It is known that the kinetic control leads to a dissimilar dissolution rate of different phases and microstructures. 35
Within the chloride concentration range from 0·05 to 0·1M, for instance, the pits on the chromium–nickel steel surfaces were of covered type and great depths. At higher chloride concentrations, the size and number of large pits for Fe–18Cr–9Ni steel grew, significantly exceeding those observed on chromium–manganese steel. The surface surrounding the pits was also subject of a general attack, but considerably weaker than that observed onto the chromium–manganese steels. The increased number of macropits as well as the feeble attack on the unaffected pits’ surface could be attributed to the increased resistance of the passive layers obtained during the anode polarisation of the chromium–nickel steel.
Intensification of the general attack on the surface was observed when the phosphoric acid concentration increased up to 40%, in presence of chloride ions. It was pronounced particularly in Cl− concentrations beyond 0·1M by selective dissolution of single grains (probably with more suitable for the dissolution crystal lattice orientation) (Fig. 9a). On Fe–18Cr–12Mn–0·6N steel, well visible pits were observed in chloride concentrations over 0·5M. These pits were of an average size of 80-100 μm and irregular shapes. For 1·0M NaCl concentration, the total surface dissolution was significant, and any traces caused by preliminary machining treatments of the samples were completely erased. The pits had irregular shapes and occupied a considerable part of the surface (Fig. 10). The surface of Fe–18Cr–9Ni steel was more weakly attacked up to a chloride concentration of 0·5M. In the presence of 1·0M NaCl, the surfaces attained microetched structures (Fig. 9).
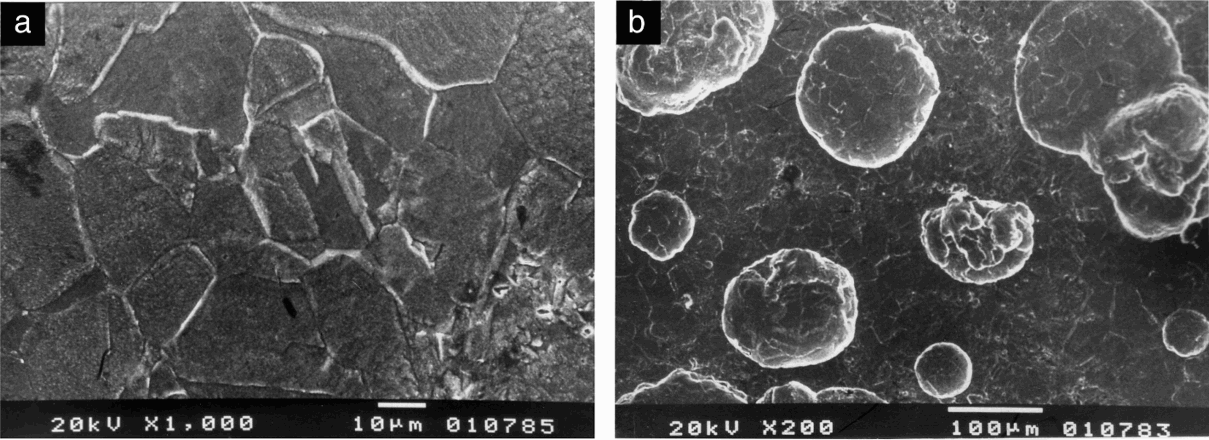
a surface out of pits; b general view of surfaceSurface of Fe–18Cr–9Ni steel samples after polarisation test in 40%H3PO4+1·0M NaCl
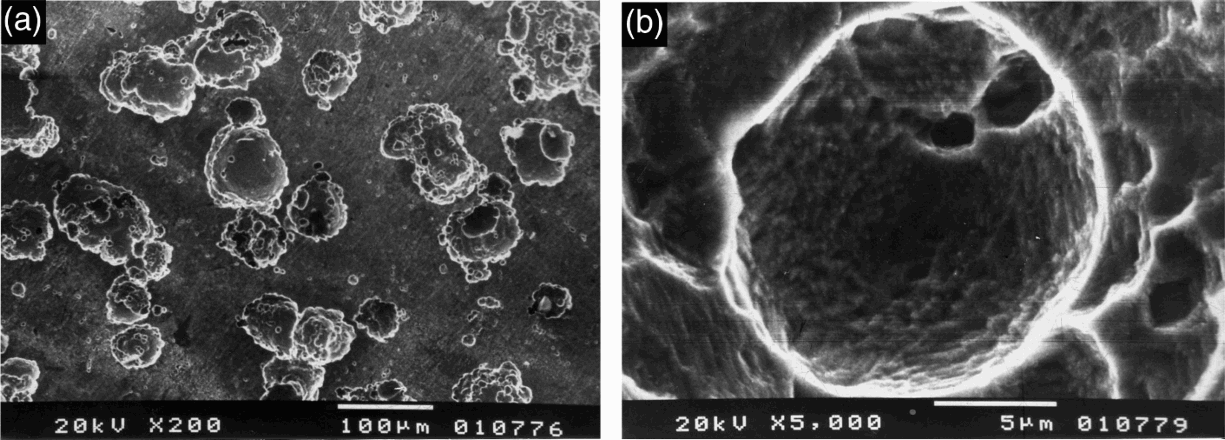
a general view of surface; b pit bottomSurface of Fe–18Cr–12Mn–0·6N steel samples after polarisation test in 40%H3PO4+1·0M NaCl
At acid concentration of 75%H3PO4, pitting corrosion was not observed even with maximum chloride concentration of ∼0·5M. The corrosion attack was general, spotted and went deeper with increase in the aggressive ion concentration, i.e. from weak etching structure at 0·05M NaCl towards strong corrosion and formation of deep caves of uneven, stepwise bottom. A small number of the grains were strongly attacked by the corrosion.
In the solution of 40%H3PO4+1·0M NaCl and anodic polarisation of samples of Fe–18Cr–12Mn–0·6N and Fe–18Cr–9Ni steels beyond 0·8 V(SCE), a green coloured compound was precipitated into the solution. In the literature background, such compound is termed as green rust and is commonly reported when mild steels are studied in phosphate solutions containing chlorides. 36 According to the authors, the green rust has different composition depending on the composition of the steel and most of all of the solution. In the experiments performed with phosphate solutions, the green rust was observed only in the presence of chlorides.
X-ray photoelectron spectroscopy analysis of passive layers on Fe–18Cr–12Mn–0·6N
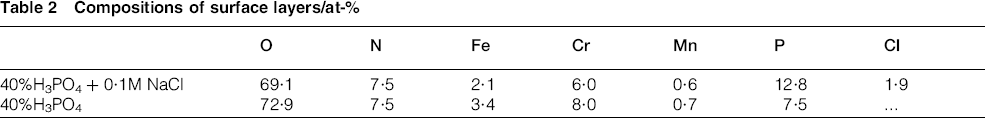
X-ray photoelectron spectroscopy analyses were carried out to see what was the chloride effect on the composition of the passive layers formed during the polarisation of Fe–18Cr–12Mn–0·6N steel samples [anodically polarised at +0·4 V(SCE)]. The tests lasted 2 h in 40%H3PO4 with and without addition of 0·1M NaCl. The data reveal that, at this anodic polarisation, the density of the passive current attains a minimum, which, in general, corresponds to most stable state of the passive layer (to some extent related to its maximal thickness). Table 2 summarises results obtained for the composition of the surface layers.
Compositions of surface layers/at-%
Several oxidation states were determined for the elements Cr, Fe, N and O, as well as their type and percentage share of the peak total areas for the respective elements, as they are presented in Fig. 11 and Table 3. Phosphorus has only one characteristic peak of its bond with oxygen, and it might be considered that the entire analysed quantity is in the form of phosphate. Therefore, the ratio (%O–4%P)/4%P represents the relation of the total oxygen quantity in the oxide, hydroxide and ads.H2O to the oxygen in the phosphate groups in the passive layer. Spectrum decomposition was not performed for the elements Mn and Cl because of their low quantities.
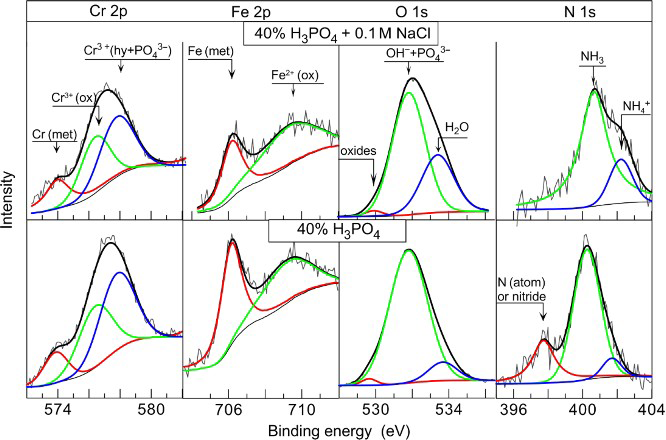
X-ray photoelectron spectroscopy spectra of Cr 2p, Fe 2p, O 1s and N 1s for Fe–18Cr–12Mn–0·6N SS anodically polarised at 0·4 V(SCE) for 2 h in 40%H3PO4 with and without NaCl
Percentage proportions of basic forms of elements Cr, Fe, O and N in passive layers formed in 40%H3PO4 with and without NaCl

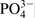 )
)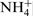
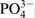 /H2O
/H2OThe data summarised in Table 2 reveal that the main differences in the compositions of the surface layers formed in 40%H3PO4, as results of the chloride action, are in the high content of phosphates and insertion of chlorine into the passive layers. The ratio (%O–4%P)/4%P considerably decreases from 1·47 to 0·35 in the presence of chlorides. Further, the amount of the water absorbed by the surface layers increases. Hence, in the solution of 40%H3PO4+0·1M NaCl, the main quantity of O is bonded to the phosphorus and the amounts of the metal oxides reduce while those of the phosphates increase.
The spectrum of the nitrogen at 397·8 eV corresponding to nitrogen in atomic state31,32 or as a nitride30,33 is missing when the surfaces were treated in 40% acid with chlorides. Despite this, the total amount of the nitrogen remains unchanged owing to the increase in the third peak at 402·2 eV, corresponding to 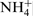 . In this context, Vanini et al.,
32
who reported that the bonds with energies of 400·2 and 402·1 eV are related to N–H (
. In this context, Vanini et al.,
32
who reported that the bonds with energies of 400·2 and 402·1 eV are related to N–H (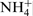 is the suggested composition), may be cited. Moreover, they state that these bounds are localised only on the external layer of the passive film. According to Fu et al.,
30
particles NH3 and
is the suggested composition), may be cited. Moreover, they state that these bounds are localised only on the external layer of the passive film. According to Fu et al.,
30
particles NH3 and 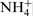 in the passive layers are as a result of the reaction of nitride or N in the steel with the solution. The absence of a peak at 397·8 eV can be explained by the increased thickness of the passive layer and consequently the impossibility of the analysing beam to reach the metal/oxide interface, i.e. the location where there is a large accumulation of nitrogen on the metal surface.
in the passive layers are as a result of the reaction of nitride or N in the steel with the solution. The absence of a peak at 397·8 eV can be explained by the increased thickness of the passive layer and consequently the impossibility of the analysing beam to reach the metal/oxide interface, i.e. the location where there is a large accumulation of nitrogen on the metal surface.
The increased ratio of Cr/Fe, and the augmented amount of H2O and 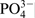 , when nitrogen is missing in either atomic or nitride states when tests were carried in 40%H3PO4+0·1M NaCl, reveals that the passive layers are of increased thickness. It might be suggested that the anodic polarisation boost the chloride ions to build an additional external phosphate layer with increased amounts of water, chlorides and ammonia.
, when nitrogen is missing in either atomic or nitride states when tests were carried in 40%H3PO4+0·1M NaCl, reveals that the passive layers are of increased thickness. It might be suggested that the anodic polarisation boost the chloride ions to build an additional external phosphate layer with increased amounts of water, chlorides and ammonia.
Conclusion
The presence of chlorides in phosphoric acid solutions of various concentrations affects unfavourably the general corrosion resistance as well as the formation and stability of the passive layers of chromium–manganese–nitrogen (Fe–18Cr–12Mn–0·6N, Fe–14Cr–15Mn–0·2N) and chromium–nickel (Fe–18Cr–9Ni) stainless steels. At applied anodic polarisation, the chlorides provoke pitting corrosion in solutions of 5 and 40%H3PO4, while in 75% acid, the irregular polishing effect was observed.
The composition of the stainless steels affects strongly the corrosion behaviour in the phosphoric acid solutions containing chloride ions. The replacement of nickel by 12% manganese and 0·6% nitrogen in Fe–18Cr–12Mn–0·6N steel reduces the general corrosion resistance and hinders the attainment of anodic passivity state. The passive region remains almost unaffected. The lower chromium and nitrogen content of Fe–14Cr–15Mn–0·2N steel compared to that in Fe–18Cr–12Mn–0·6N steel deteriorates further all corrosion characteristics: this is manifested by higher values of the corrosion rate, critical and passive currents as well as by increased pit nucleation at lower degree of anodic polarisation.
In general, it can be stated that the usage of the three examined steels in phosphoric acid solutions with concentration above 5% containing chloride over 0·01M is not acceptable because of the great uniform corrosion rate and pitting corrosion development. Implementation of anodic protection in these environments is inadmissible because of high risk of localisation of corrosion attack.
References
 ,
, 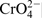 ,
, 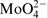 and
and  anions’
anions’