
Abstract
Interest in hydrogel materials is growing rapidly, due to the potential for hydrogel use in tissue engineering and drug delivery applications, and as coatings on medical devices. However, a key limitation with the use of hydrogel materials in many applications is their relatively poor mechanical properties compared with those of (less biocompatible) solid polymers. In this review, basic chemistry, microstructure and processing routes for common natural and synthetic hydrogel materials are explored first. Underlying structure–properties relationships for hydrogels are considered. A series of mechanical testing modalities suitable for hydrogel characterisation are next considered, including emerging test modalities, such as nanoindentation and atomic force microscopy (AFM) indentation. As the data analysis depends in part on the material’s constitutive behaviour, a series of increasingly complex constitutive models will be examined, including elastic, viscoelastic and theories that explicitly treat the multiphasic poroelastic nature of hydrogel materials. Results from the existing literature on agar and polyacrylamide mechanical properties are compiled and compared, highlighting the challenges and uncertainties inherent in the process of gel mechanical characterisation.
Introduction
Hydrogels are network polymeric materials in which the polymer chains are very hydrophilic, such that they can associate with large quantities of water without dissolving. The water can be tightly bound to the polymer network or free to move within the polymer network. 1 Because of the large water content, hydrogels are quite biocompatible, and can be made with water contents quite similar to those of biological tissues (∼70%) or much greater (up to and beyond 99% water). They were first used for soft contact lenses in the 1960s 2 and have since been adopted for use as catheter coatings, 3 wound dressings, 4 and in drug delivery applications. 5,6 Current research for future uses of hydrogels indicates that they have great promise in tissue engineering, cell encapsulation, nanoparticle coatings, and in diagnostic microdevices such as microfluidics and MEMS devices. 1,7–10 Hydrogels have also increasingly been used in basic science biological studies investigating cell–material interactions, 8 including those in which gel mechanical properties are varied systematically in order to study mechanical influences on stem cell differentiation. 11–14 The mechanical behaviour of hydrogels has long been recognised as fundamentally important, and previous reviews have highlighted different aspects of hydrogel mechanics without addressing all of the topics that will be considered here. 12,15,16
Because a substantial fraction of hydrogel materials is water, a limiting factor in the widespread adoption of hydrogels for biomedical applications has been their poor mechanical properties. 7 This is particularly the case when hydrogels are compared with traditional engineering materials in the context of mechanical behaviour. As these are materials that have relatively small elastic modulus values and which exhibit neither the behaviour of something solid nor something liquid, both the measurement and interpretation of mechanical data presents significant challenges for the researcher. First, it can be difficult to ‘grip’ the samples for testing. Second, the elastic modulus of most hydrogels is on the order of kiloPascals (kPa), while most mechanical testing equipment is optimised for the range of MegaPascals (MPa) to GigaPascals (GPa). Further, since hydrogels are multi-phase materials consisting of a porous ‘solid’ with a liquid (water) solvent phase, full understanding of the mechanical response does not result from simple treatments established for solid polymers, and more complex analysis is required. All of these issues must be addressed for researchers working to develop more mechanically robust hydrogel materials, and all will be addressed in the course of this review.
Hydrogel chemistry, preparation and microstructure
Hydrogels can be categorised according to the nature of the polymer network, including the origin of polymeric material, basic polymer chemistry, details of preparation and types of chemical bonding present in the final network. 17,18 Hydrogel polymer network origins fall into three main categories, those of synthetic polymer, those of natural polymer and hybrid materials with both synthetic and natural elements. 10,17 Synthetic polymers (Figure 1a–d ) that have been commonly employed in hydrogel studies include include poly(ethylene oxide) [PEO although sometimes poly(ethylene glycol), abbreviated PEG, when the molecular mass is below 20 000 g mol−1], poly(2-hydroxyethyl methacrylate) (polyHEMA), poly(vinyl alcohol) (PVA) and poly(acrylamide) (PAAm). 19–28 Natural polymer (Figure 1, e-f) hydrogels include those of proteins, such as collagen 29 and silk, 30 denatured proteins, such as gelatin, 31–35 and polysaccharides, such as agar 36–42 and alginate. 43,44 Both synthetic and natural gels can be multicomponent, either as grafted block copolymers or as interpenetrating networks of two or more independent polymers. Degradable linkers have been incorporated into a chemically stable gel for controlled release or regenerative medicine applications. 6 Another mechanism of generating hybrid multicomponent materials is the grafting of a biological recognition motif, such as the RGD (arginine–glycine–aspartic acid) cell recognition sequence found in many cell adhesive proteins such as integrins, 45 to a synthetic polymer in order to improve cellular biocompatibility with the gel network.
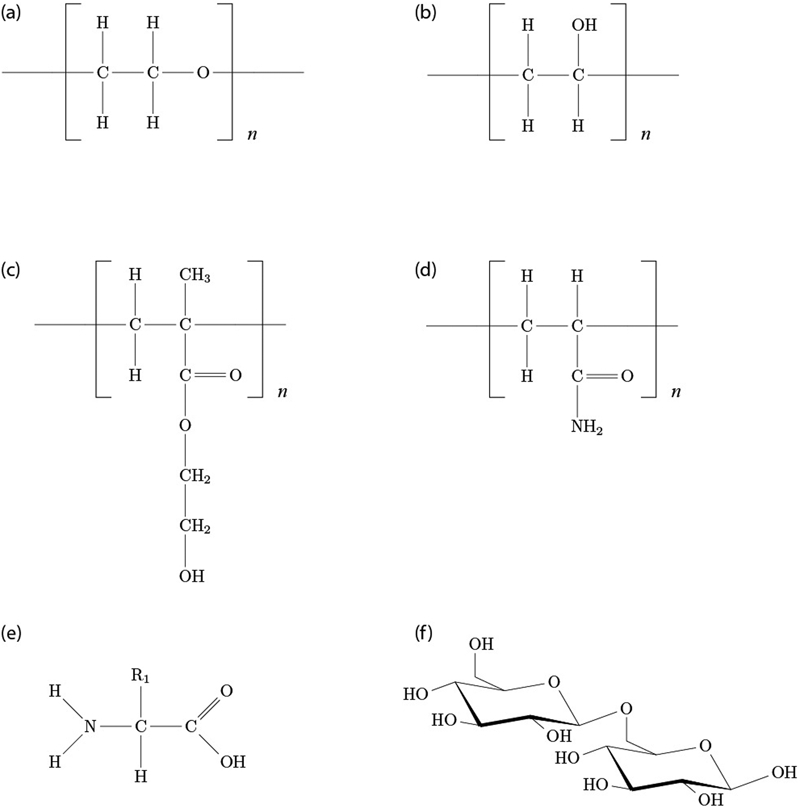
Chemical structures for some common hydrogels
In terms of determining the physical properties of gels, the most important distinction is that between polymer networks that are chemically bonded and those that are networks of physical entanglements between polymer chains. Figure 2 illustrates a series of chemical (Fig. 2a–c ) and physical (Fig. 2d–f ) hydrogels. For chemical gels, there are covalent cross-links at intersection points of polymer chains, and in which the network can be (1) ideally chemically bonded; (2) non-ideally chemically-bonded, with polymer chain self-loops and free ends or (3) a double network gel, in which there are two distinct networks, each only covalently linked to ‘like’ chains and forming an interpenetrating network structure. For physical gels, there are effective cross-link points which can have a range of sizes of the region connected by non-covalent bonds. In the simplest case, chains loop around other chains and form physical entanglements (Fig. 2d ). In both Fig. 2e and f , the regions of polymer chain engagement at the effective cross-link points are larger than at a single covalent point or a single chain entanglement; in Fig. 2e , the formation of helices is shown, and Fig. 2f represents the algae based gel alginate, in which divalent cations (most commonly Ca2+) form complexes joining the guluronic acid domains in the polysaccharide chains. 44 The nature and definition of the cross-link points is fundamentally distinct when comparing chemical and physical gels, related to the relative ease of shifting these cross-link points under applied mechanical loading. Chemical cross-links are relatively immobile compared with physical cross-links. Chemical and physical gels can be produced from the same base monomer chain, as has been shown in comparing gelatin 32 or alginate 44 gels with different bondings.
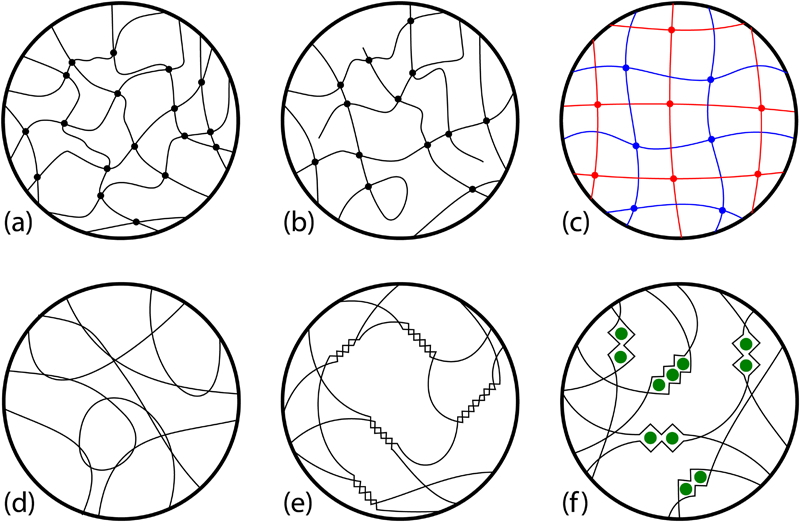
Schematic microstructures of gels to define types of cross-linking
Hydrogels can be considered as molecular scale porous materials in which the pore space is occupied by water. 1 In a strict definition of a hydrogel, first the chemical network is formed and then the network is swollen in water, 5 although sometimes the network formation also takes place in an aqueous environment. For chemical gels, cross-linking methods include chemical reaction and photopolymerisation with ultraviolet light or by irradiation; physical cross-linking methods include thermal, as in setting from a melt, or from a reversible ‘bridging’ reaction such as in the divalent Ca2+ bridges in alginate gels, which are not covalently linked (Fig. 2f ). 1
Swelling behaviour in hydrogels is important both dynamically and at equilibrium. (poroelastic relaxation experiments, described later in the section on ‘Poroelastic’, have at times been called deswelling experiments, as they involve the movement of water out of the polymer network under physical stress.) A volume swelling coefficient (Q) is defined as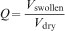
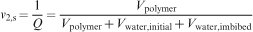
Another key parameter in understanding fundamental scaling laws in the context of hydrogels is the (number) average molecular weight between two adjacent cross-linking points 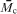
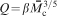
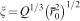
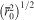
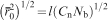
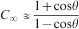
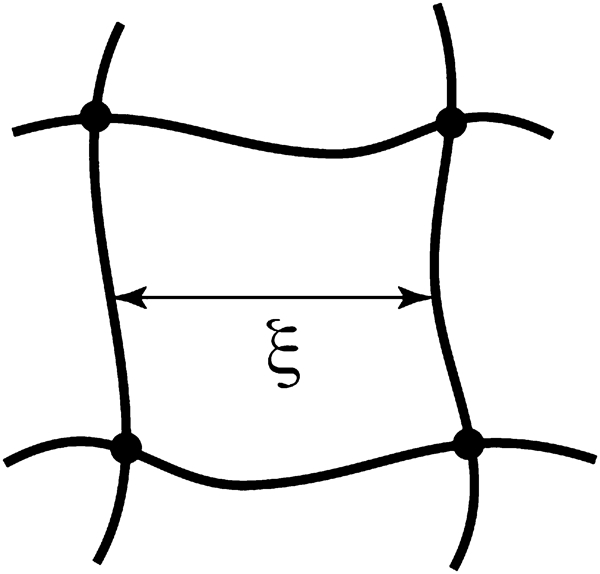
Definition of hydrogel mesh size ξ, the distance between effective cross-linking points

A scanning electron microscope (SEM) image of a 15% gelatine mixture quenched from molten in liquid propane and freeze dried prior to imaging. The specimen preparation is unlikely to result in an identical microstructure to the one that results from the slow cooling of the hydrated molten mixture as in typical gelation. The scale bar is 2 μm in length
Any of the structural parameters introduced here (v
2,s, Q, 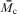
Hydrogel mechanical characterisation techniques
The basic mechanical testing techniques and apparatuses used for polymeric materials more generally are also used for mechanical characterisation of hydrogels. As with most polymers, hydrogels exhibit time dependent mechanical behaviour due to intrinsic viscoelasticity of the polymer network, but with an additional time dependent deformation mechanism due to fluid flow. Thus, time factors heavily in the planning and execution of mechanical experiments on hydrogels, which can be characterised in either the time or frequency domain. Here we will consider the six most common testing techniques (Fig. 5): tension, compression, which can be either unconfined or confined, local indentation with a probe and frequency based tests such as shear rheometry or dynamic mechanical analysis (DMA). Finally, there are some non-contact mechanisms used to infer mechanical properties.
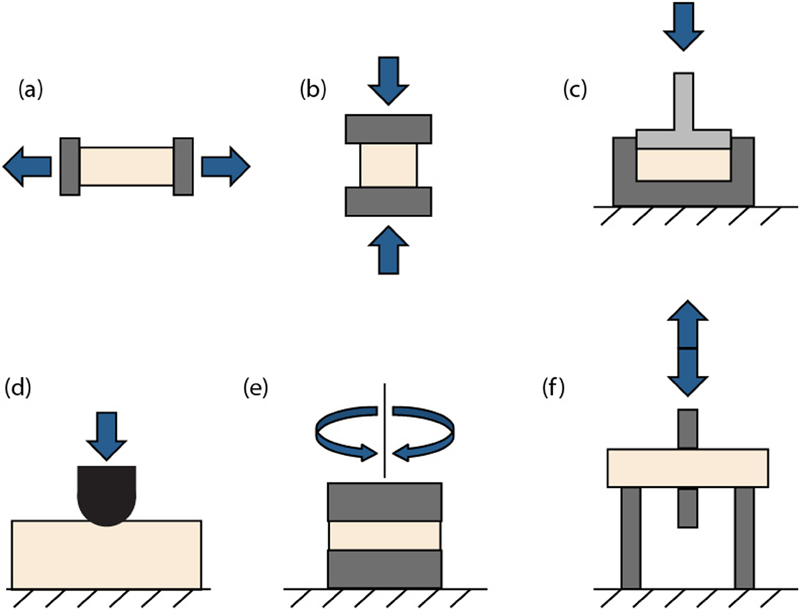
Schematics of different types of mechanical testing set-ups used for testing hydrogel mechanical properties
Universal Test-frame
The most common tool used for mechanical characterisation of materials is a Universal Test-frame, which is capable of carrying out a wide variety of experimental tests. Most frames are uniaxial, in that motion is actuated and force measured along a single axis, although biaxial instruments have become increasingly common in recent years. 49 The mechanism of motion actuation can vary, including servohydraulic systems, electromechanical systems (both very common) and systems driven by speaker coils (less common). The same systems can be used for tension and compression testing, with different hardware at the point of sample contact. For tensile testing, key to execution of effective tests is good sample gripping, something that is particularly difficult when the specimens are compliant and hydrated, as in the case of hydrogels. Strategies for addressing this challenge include the use of cardboard tabs, double sided tape and glue to assist in the gripping the hydrogels. 39 For compressive testing, the sample can either be unconfined (Fig. 5b ) and compressed between two non-porous platens, 26,39,40,50 or confined (Fig. 5c ) to a container and compressed with a single porous platen. 51 The latter case is a testing modality unique to multiphase materials, as the motion of fluid out of the sample, through the porous platen, is expected (this will be discussed further in the section on ‘Poroelastic’). Universal Test-frames range in size from extremely large (meganewton, MN) scale machines used in testing civil engineering components to small modern machines optimised for soft biological materials or biomedical components including hydrogels. A typical load cell for such testing would have a maximum load capacity of only 5 or 10 N.
For both tension and unconfined compression testing, the load–displacement (F–x) data are converted to stress–strain (σ–ϵ) data using simple geometrical relationships 52 and the Young’s modulus E and failure strength σ f are reported. Typical tests are done at fixed rate until specimen failure, and due to the time dependent nature of hydrogels, testing at a variety of strain rates may be required (Fig. 6a ). 53 Especially given the nature of the time dependent response of hydrogels, in addition to simple tests at fixed rate, the specimen may be subjected to creep (extension at fixed load or fixed stress) or relaxation (load decrease at fixed deformation or strain) experiments where variation in response with time is of interest and the loading is fixed (Fig. 6b ).
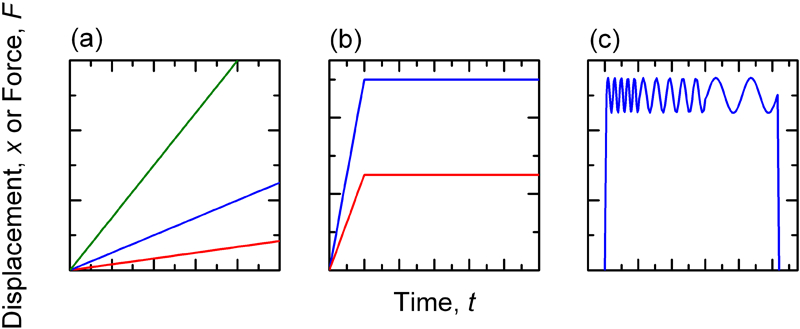
Mechanical test types: imposed displacement–time (x–t) or force–time (F–t) for a fixed rate for varying rates, b static creep or relaxation at fixed force or displacement and c sinusoidal loading at varying frequency
Indentation
In indentation testing, a probe of known geometry is brought into contact with a material surface, pressed into the material, and retracted again. Historically, indentation testing was the gold standard for examining the hardness of metals,
54
and indenters were dead weight loading devices coupled with optical microscopes for examination of the residual indent impression. However, the growth in recent years has been in depth sensing indentation, in which the full indentation load–depth–time (P–h–t) profile is recorded and analysed for material property deconvolution. Thus, the indentation test is a local version of a compression test, where the sample is compressed in a small region instead of across the entire surface. The most commonly reported parameter associated with indentation testing is the reduced modulus E
R, which for compliant materials is equivalent to the plane strain modulus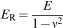
Minimal sample preparation is required for indentation testing. As a result, indentation has been growing in popularity for testing materials, such as hydrogels, which would be difficult to ‘machine’ into a regular specimen geometry. Hydrated, compliant specimens can also be difficult to ‘grip’ for tensile or compressive mechanical testing, which is not required in probe based testing. The relatively small volumes of material required for quantitative indentation testing means that valuable samples can be examined, and high throughput screening methods may prove useful for materials selection. Further, inhomogeneous graded or patterned gels can be probed locally for quantification of the local inhomogeneity, using a mapping technique across the sample surface. 55 Finally, it is straightforward to keep hydrogel samples hydrated throughout the duration of an indentation test.
Indentation testing has been used to study hydrogels at large (mm) 40,56,57 and at small (nm to μm) length scales. 13,19,22,27,41,58,59 The technique is highly adaptable with regard to length scale, as the probe geometry and contact area can be tuned to optimise the testing to the instrument such that the forces and displacements are appropriately scaled in terms of both range and resolution. 60,61 At large length scales, a Universal Test-frame can be adapted for execution of an indentation test. More recently, ‘nanoindentation’, or indentation testing at small length scales, has emerged as a leading technique for the mechanical investigation of a wide range of materials. 62,63 For this small scale indentation, there are different approaches to take with respect to the testing instrumentation. There are a number of commercial stand alone nanoindentation systems that have been developed for small scale quantitative testing and that have been used for hydrogel characterisation. 19,22,58,59 For historical reasons, these instruments are largely optimised for testing stiff engineering materials, and adaptations of typical test techniques (such as the use of larger indenter and contact radii) are employed for testing compliant polymers and hydrogels. An alternative approach is to utilise atomic force microscopes (AFMs) for gel indentation testing. 12,13,27,41 The primary difference between commercial nano indentation systems and AFMs for indentation testing lies in the different physical principle governing transducer operation: commercial nanoindenters utilise speaker coils or capacitance gages to directly actuate the indenter probe into the sample, while AFMs actuate the tip indirectly via a calibrated cantilever. The cantilever based systems are thus easy to optimise for testing different classes of materials with different stiffness simply by switching the cantilever stiffness. However, the quantitative nature of the test results is extremely sensitive to cantilever calibration. In both cases, feedback control is required for quantifying time dependent deformation. In practice, neither nanoindentation nor AFM indentation utilising existing commercial equipment is ideally suited for hydrogel testing but both are likely to grow in popularity in coming years as the need for gel property quantification at small length scales continues to increase.
Frequency based testing
Frequency based sinusoidal testing is commonly carried out using ‘black-box’ testing systems (Fig. 5e and f
), using rheometry in shear
20,43,64,65
or in a range of DMA test modes including shear, tension or bending.
51,66
The sample, often in a geometry specified for the instrument and test type, is loaded into the instrument and different types of ‘sweep’ measurements can be performed. Most common is a frequency sweep (Fig. 6c
), where the sinusoidal oscillation is at a fixed (and usually quite small) strain but the oscillation frequency is varied. The test output is the storage (G′) and loss (G″) components of the complex modulus G* as a function of frequency, where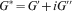
New developments have expanded the use of frequency based techniques beyond traditional rheometry or DMA. Sinusoidal indentation testing utilising frequency sweeps has gained some recent traction for polymer and hydrogel characterisation 41,59,67 and dynamic confined compression measurements have been used, 51 for comparison with traditional confined compression creep measurements. For small scale dynamic measurements, a novel technique termed ‘microrheology’ has emerged, in which tracer particles are embedded within a gel and their motion monitored. 12 This allows for local probing of viscoelastic behaviour throughout a gel’s thickness.
Non-contact
The techniques described above have all involved physically extending or compressing a sample. There are some additional mechanisms for measurement of physical properties without substantially deforming the sample. Permeability can be measured by flowing fluid through a gel at known rate and measuring pressure with a pressure transducer 20 or by varying the pressure and measuring the flowrate. 68 This type of measurement is sometimes denoted the ‘direct permeability’ since no material model is required to interpret the measurement. Other measures rely on indirect mechanical estimates of mechanical properties based on the pioneering models of Flory and Rehner in the 1940s. 47,48 The actual measurements can come from Gel Permeation Chromatography (GPC) 69 or from light scattering. 70–73
Models for interpretation of mechanical results
Once experimental data have been collected, the load–displacement–time (F–x–t, or P–h–t for indentation) or stress–strain–time (σ–ϵ–t) data must be interpreted within some mechanical framework in order to obtain mechanical property values. The simplest case, for linearly elastic and isotropic materials, is when the slope of the linear, small strain, stress–strain (σ–ϵ) curve from a uniaxial (tensile or compressive) test is taken as the elastic modulus E. For hydrogels, the fundamental constitutive response is more complicated, and as such more complicated analyses must be considered, including elasticity under large strains and the presence of time dependent deformation. Both analytical and numerical computational approaches will be considered here.
Elastic and hyperelastic
The polymer network in hydrogels is often treated using concepts originally developed for the study of rubber elasticity. This is a particularly good approximation for highly cross-linked chemical gels (Fig. 2a
) in which the polymer network is in effect a single molecule due to the high connectivity. For such a rubber-like network, entropic elasticity is dominant, and the elastic modulus G (note this is the terminology utilised in the rubber elasticity literature, and is not necessarily meant to indicate shear modulus) in the unswollen state can be simply expressed using the theory of rubber elasticity
15,47,48,74,75
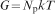
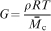
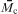
For the swollen state, characteristic of a hydrogel with volume swelling ratio Q, the solvent swollen modulus is the product of the unswollen (pure polymer) modulus and the linear swelling ratio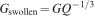
For simple uniaxial extension or compression, the relationship between true stress τ, and stretch, 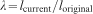
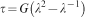
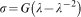
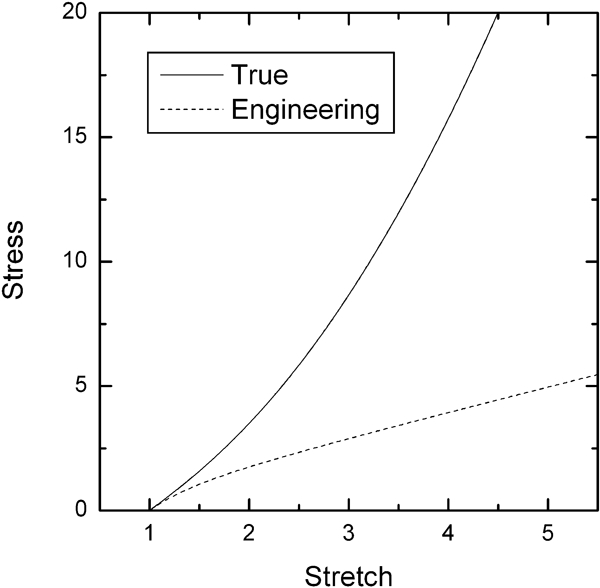
Stress–stretch relationships for rubber elasticity, illustrating the tensile relationship for both true stress and engineering stress (equations (10) and (11) respectively)
The elastic modulus is an equilibrium parameter, and to this point we have considered only elastic deformation. An approximate equilibrium modulus is measured with the use of relatively slow strain rates in tensile or compressive testing in many studies of hydrogel mechanical behaviour. However, there is intrinsic time dependent deformation present in hydrogels due both to the viscoelasticity of the polymer network and due to the motion of fluid within the gel, and there is thus an opportunity to gain additional structural information about a gel via the examination of this dissipative deformation. Thus, we will next consider hydrogel non-equilibrium behaviour.
Time dependent
Time dependent mechanical behaviour is fundamental to the physics of polymers, including hydrogels. The treatment of this time dependence ranges from relatively simple empirical measurements through to complex models treating the solid and fluid phases and their interactions explicitly using the coupled equations of poroelasticity. Here, viscoelasticity will be considered first, followed by poroelasticity.
Viscoelastic
The basic premise of viscoelasticity is that it parallels the framework of elasticity but with time dependent functions replacing the elastic constants.
77
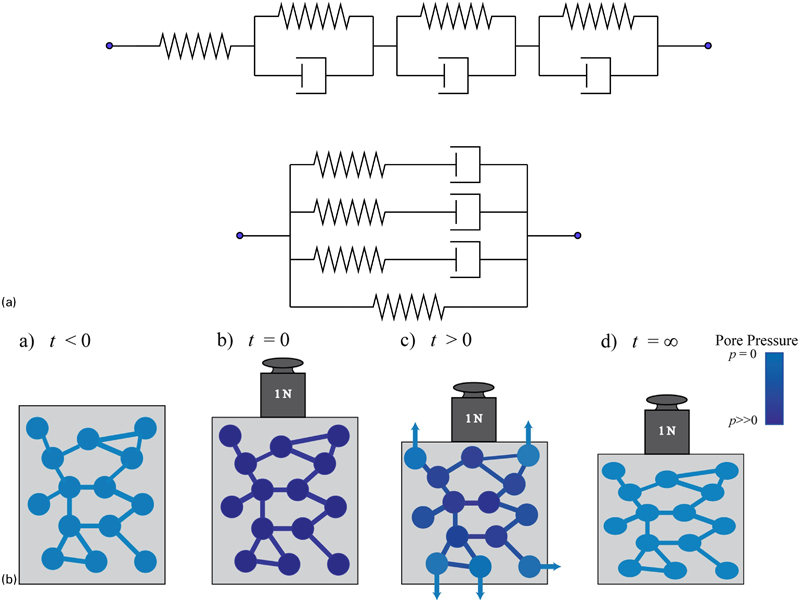
It is quite typical to assume that that volumetric deformation is time independent, and there is a solitary time dependent shear function G(t). For very simple cases, physical lumped parameter models (Fig. 8a
) are used and explicit spring and dashpot coefficients are reported. However, more generally, polymer viscoelasticity in the experimental time domain is treated with the assignment of an empirical Prony series (summed exponential function) to describe the material’s time dependent constitutive response, for example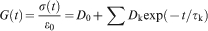
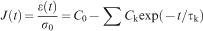
Time dependent deformation mechanisms
Further, viscoelastic experiments can be conducted in the frequency domain, as in the case of DMA 66 and rheometry. 64 Spring and dashpot models can be used to interpret the data, or more sophisticated approaches such as fractional derivative models can be employed. 66 Viscoelastic textbooks 77 show how data can be converted between the frequency domain and the time domain, as there are not different physical functions describing viscoelastic behaviour in the frequency versus time domain but a single physical viscoelastic response for the material. However, in practice, it is relatively infrequent to find the conversion done routinely. DMA and rheometry data are frequently reported as a plot of storage and loss moduli (G′ and G″) versus frequency (ω) without further analysis. To date, there has been relatively little success in trying to assign any of the viscoelastic coefficients to explicit physical processes or to derive expected values from first principles based on polymer structure.
Poroelastic
The time dependence demonstrated by hydrated materials has been described using multiphasic poroelastic models. Poroelastic
78
(also called biphasic
79
) constitutive behaviour is when time dependence arises due to the flow of a fluid through an elastic (or viscoelastic), porous solid (Fig. 8b
). The material properties of interest in poroelastic material characterisation include the elastic properties of the porous skeleton: the (shear) elastic modulus G and the (drained) Poisson’s ratio v, the undrained Poisson’s ratio v
u and a parameter α which ranges from 0 to 1 and describes fluid-solid interactions. The fifth constitutive parameter is the Darcy permeability κ which is a function of the fluid viscosity η and the intrinsic permeability k = κη. The permeability can be thought of as analogous to the diffusion coefficient, but under the motive of a mechanical, rather than a chemical, gradient. The intrinsic permeability is also indicative of the effective pore size (ξ) of the porous skeleton k = Cξ
2 where C is a constant. A frequently reported parameter for poroelastic data analysis is the aggregate modulus H
A
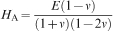
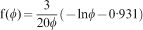
Poroelastic data analysis is complicated by the fact that the governing equations are coupled, and as such there are relatively few types of mechanical tests for which simple, closed form solutions exist and can be fit directly to data. The exception is the case of one-dimensional consolidation, or confined compression (Fig. 5c ). For both the assumption of a step load or a constant rate ramp followed by a hold period at constant load (stress), relatively simple solutions exist and are in the form of summed exponentials. 83 However, for more complicated loading schemes, such as indentation testing, numerical or computational models are often required. Several recent papers have considered the measurement of poroelastic properties, particularly the elastic modulus and hydraulic or intrinsic permeability, of hydrogels with good success using indentation techniques. 19,44,83–87 Permeability has been measured directly by flowing fluid through a gel, and there was good agreement between direct permeability measurements and values obtained via indentation testing. 19,88 However, compared with elastic modulus values, the permeability is less frequently measured and reported for hydrogel materials.
Compiled mechanical results for common hydrogels
Next, material property results from a number of studies will be compared as a function of gel polymer concentration for two common hydrogel systems, one physical gel and one chemical gel. Where tabular data were unavailable, data were digitised using open source software. 89
Agar
Agar gels are largely made of the linear polysaccharide molecules that form from the disaccharide agarose. They are physical gels that form on cooling, with a gel temperature near 35 C. 39
Elastic modulus values for agar gels have been compiled from a number of sources
39–41,51,56–59
and are plotted as a function of gel concentration in Fig. 9. Owing to the large range in values, and the uncertainties associated with assuming Poisson’s ratio values, the data are plotted as reported, without attempt to convert any reported shear (G), storage (E′ or G′), reduced (E
R) or aggregate modulus (H
A) to a Young’s modulus (E) value. As can be seen from the plot, for any single value of the gel concentration, the reported modulus values vary by up to three orders of magnitude. There are factors that are likely confounding the simple plot, such as agarose chain molecular weight.
39
However, within any single set of data, the scaling with gel concentration is consistent with either of two predicted concentration dependencies proposed in the literature, either quadratic
90
or in concentration to the 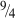
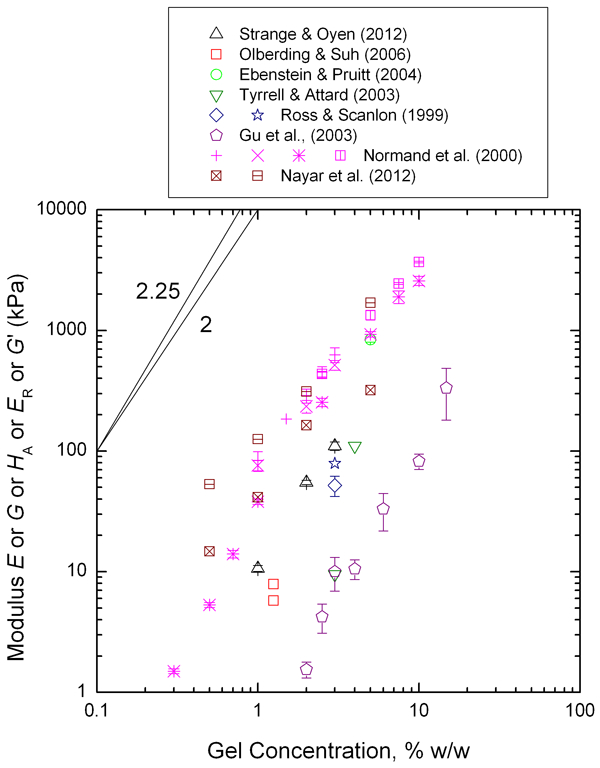
Plot of compiled agar gel elastic modulus values as a function of gel concentration. Scaling laws for modulus with polymer (gel) concentration according to cellular solids (power law 2 90 ) and polymer physics (power law 2·25 91 ) are shown for reference. Data compiled from Refs. 39–41, 51 and 56–59.
Data for hydraulic permeability of agar gels as a function of gel concentration are much more consistent than modulus data across studies,
51,56,57,92,93
as shown in Fig. 10. As the polymer concentration increases, the permeability decreases, implying that the effective pore size is also decreasing. However, the data do not seem to indicate a simple change in pore size at fixed fibre size, as might be expected. The trend is steeper than would be predicted from a −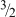
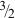
Plot of compiled agar gel permeability values as a function of gel concentration. Data compiled from Refs. 51, 56, 57, 92 and 93
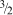
To summarise the agar data (Figs. 9–11), the modulus increases and the intrinsic permeability decreases with increased polymer content, or conversely the modulus decreases and intrinsic permeability increases with increased water content. There is substantial scatter in the modulus data, and less scatter in the permeability data. The modulus data seems to agree well with predicted behaviour from scaling arguments, but permeability does not. This may be because agar is more appropriately treated as poroviscoelastic rather than poroelastic, which was only considered in one of the cited studies. 56 Recent work has confirmed that agar is poroviscoelastic, and that with multiple experiments the contributions from different time dependent deformation mechanisms can be decoupled. 94 To establish the generalisability of the agar results here, a second gel is next examined using the same methodology, allowing for consideration of bond type in contrasting the physical agar gel with a chemically cross-linked gel.
Polyacrylamide
Polyacrylamide (PAAm) hydrogels could be considered to be the best characterised of all gels, being ubiquitous across biomedical research, including being used in biology for gel electrophoresis to separate proteins by size and charge. 95 As with many polymer systems, the acrylamide monomer is extremely toxic in its unpolymerised form but benign once polymerised. For this reason, much effort is usually undertaken to allow for outward diffusion of unpolymerised monomer prior to using the gels for a wide range of biomedical applications. The monomer is polymerised in aqueous solution in the presence of small amounts of bifunctional cross-linker, frequently N,N′-methylenebisacrylamide (BIS or bis-acrylamide), resulting in an acrylamide network with BIS links. This reaction occurs via a free-radical mechanism, where ammonium persulfate (APS, N2H8S2O8) is used as an initiator, and a small volume of TEMED (N,N,N,N-tetramethylethylenediamine) is used to stabilise the reaction. This is typically held at fixed concentration, such that there are two key parameters determining the overall nature of the hydrogel. These have a reasonably consistent nomenclature used across the literature as follows: T represents the total polymer concentration (w/v) where this is the sum of the monomer and BIS; C is the percentage (w/w) of the polymer that is cross-linker. Thus, %T = 100%[BIS (gm)+monomer (gm)]/100 mL solution and %C = 100%[BIS/(BIS+monomer)] (gm/gm). Because the cross-linker is typically present in much smaller amounts than the monomer, it is common for the polymer concentration %T to be calculated based on the monomer only, and for the cross-linker concentration %C to be calculated from the ratio of BIS to monomer.
The relationship between pore size and both total polymer concentration and cross-linker concentration is complex. When %T is fixed, the pore size decreases with increasing %C up to a point, but then larger %C causes a more macroporous structure due to polymer chains agglomerating. 69 When %T is varied at fixed cross-linker concentration, the pore size decreases with increasing %T. Thus, a full picture for the mechanical behaviour of PAAm hydrogels requires a large data set with a range of both polymer and cross-linker concentration.
Polyacrylamide elastic modulus values (E,G or G) have been compiled from a range of sources
19–23,26,27
and are plotted in Fig. 12 as a function of total polymer concentration (%T). As was described above for the agar gels, different moduli were plotted on the same axes without conversion, as the differences between these for a single material are small compared with the scale extending over seven orders of magnitude in modulus. Two scaling lines, one corresponding to modulus going as the square of the concentration
90
and the second using the 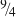
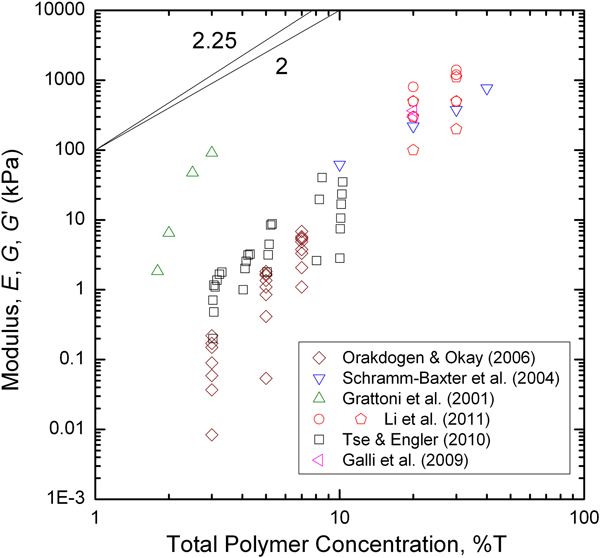
Plot of compiled acrylamide gel elastic modulus values as a function of the total polymer concentration (%T). Scaling laws for modulus with polymer (gel) concentration according to cellular solids (power law 2 90 ) and polymer physics (power law 2·25 91 ) are shown for reference. Data compiled from Refs. 19–23, 26 and 27
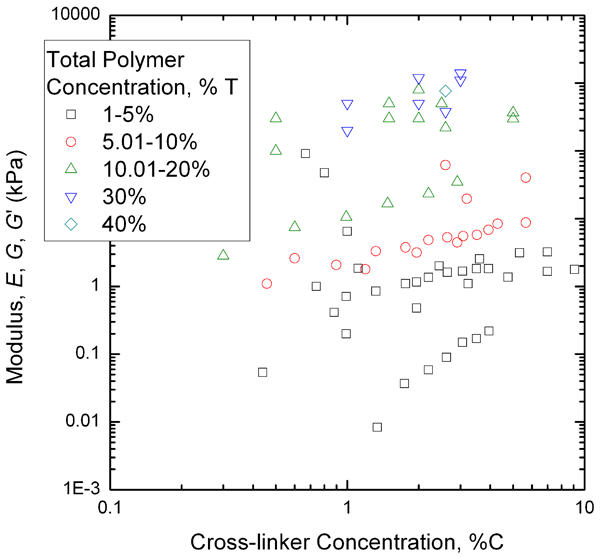
Acrylamide gel elastic modulus values from Fig. 12, replotted as a function of cross-linker concentration (%C) for data grouped according to the total polymer concentration (%T)
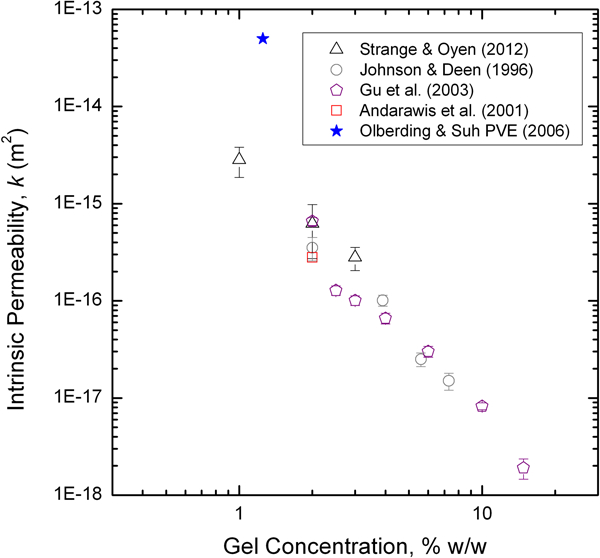
Intrinsic permeability values for PAAm gels, assuming a linearly poroelastic response, have been compiled from a number of sources where measurements were made
19,20,68,73,96,97
and one study for which permeability is here calculated as the square of the estimated pore size from GPC measurements,
69
and are plotted in Fig. 14 (note that it was assumed that the y-axis scale for Fig. 8 in Tokita and Tanaka’s work
73
is mislabeled, based on comparisons with their Figs. 6 and 7, and the data have been shifted accordingly). There is good agreement between the computed values
69
and the measured values for some of the studies,
19,68,73,96
but the data from two other studies
20,97
are substantially different, namely, larger by several orders of magnitude. The data that all agree are also consistent with a roughly −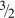
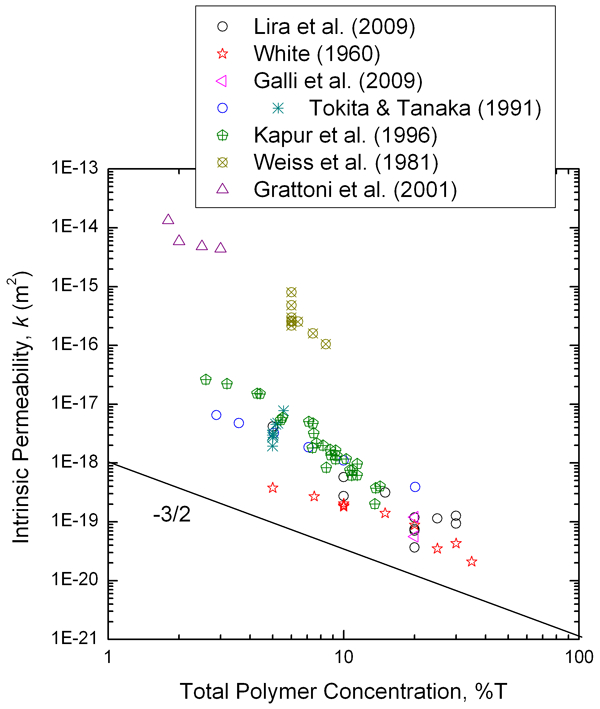
Plot of compiled acrylamide gel permeability values as a function of total polymer concentration (%T). In contrast with the agar data (Fig. 11), the data taken as a group do appear to follow a -3/2 scaling law for permeability with gel concentration. 73 Data compiled from Refs. 19,20, 68, 69, 73, 96 and 97
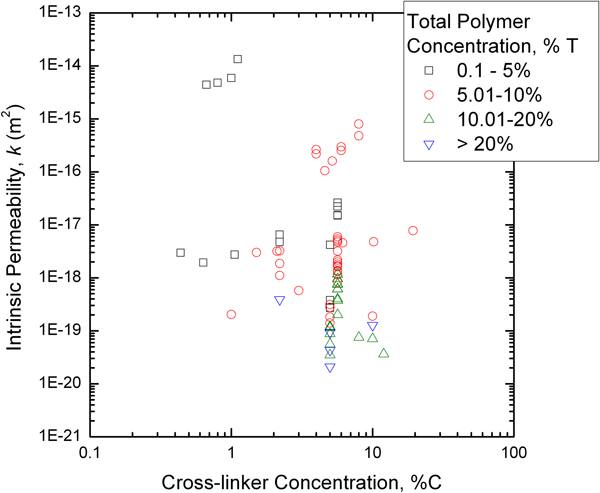
Acrylamide gel permeability values from Fig. 14, replotted as a function of cross-linker concentration (%C) for data grouped according to the total polymer concentration (%T)
Conclusion and outlook
In this review, two complementary aspects of the mechanical properties of hydrogels have been considered. For many applications, the hydrogel properties are intrinsically important, and in many structural applications, they present a major limitation for utilisation in real world applications. Mechanical measurements are further important for inferring difficult to measure hydrogel structural characteristics such as mesh size. Both of these aspects are important moving towards the introduction of hydrogels into clinical applications, such as in tissue engineering applications. Avascular connective tissues, such as the spinal disc 57 and cornea 98 are obvious targets for tissue engineering due to their relatively simple microstructures, but in both cases, the tissues are subject to mechanical loads in vivo. If hydrogels are to succeed as tissue engineering scaffolds, their mechanical properties must be sufficient and robust and repeatable methods for measuring these properties must be established.
In compiling sets of data from the literature, variability in reported elastic property values (E, E R, G, G′, H A) for the same hydrogel compositions has been shown to be quite large at any given gel concentration. For example, reported modulus values for a 2% agar gel range from just over 1–300 kPa (Fig. 9). For a 3% acrylamide gel values ranged from 10 Pa to 100 kPa, although the 100 kPa value is perhaps an outlier (Fig. 12). With that value excluded, a two order of magnitude range remains, from 10 Pa to 1 kPa. These comparisons in Figs. 9–15 show significant but not systematic variation between different studies, in which different testing modalities were utilised. This raises the question as to whether there is consistency when multiple test modalities have been compared within the same study. Good quantitative agreement between frequency based rheometry and DMA results has been demonstrated for dextran methacrylate hydrogels. 99 In one study where compression and indentation testing were used for the same 3% agar gel, the indentation measurements of modulus were 52% larger than for compression testing. 40 Indentation data, both nanoindentation and microindentation, also gave larger modulus values than either compression or DMA testing for 20% acrylamide gels, and there was only limited agreement between time and frequency based test methods. 19 A third study also compared compression and AFM indentation of hyaluronic acid gels and found no systematic variation between the test modalities with gel concentration; at small gel concentrations the indentation values were smaller, whereas at large gel concentrations the indentation values were larger. 13 These results taken together highlight potential challenges for the field. Uncertainties for the measurement of elastic modulus values in structural engineering materials are typically on the order of a few percent, not a few orders of magnitude. Significant questions remain as to both the accuracy and precision of the modulus measurements for these compliant hydrogels. Taken as a whole, the data illustrate that more studies are needed in which mechanical testing modalities are systematically compared for the same gels, in order to establish best practices for both experimental protocol and data analysis methodology.
The gels considered here, particularly those simple gels such as agar and polyacrylamide, were single component, in bulk form and subject to bulk mechanical characterisation. The modulus and permeability both scaled with gel concentration in a manner largely consistent with theoretical predictions. 73,80,90,91 Modulus increased with increasing polymer fraction (and decreased proportion of water) and permeability decreased with increasing polymer fraction (and decreased water fraction). There is thus limited scope for dramatically improving hydrogel properties in these simple systems. Recent attempts have aimed at designing unique microstructures in which modulus and permeability are decoupled. 100,101 Composite gels have been made, including gel–gel systems, such as agar–gelatine, 57,102–105 agar–collagen 106–108 or gelatine–PAAm. 109 Gels have also been reinforced with a non-gel phase, such as particles of hydroxyapatite, 110 clay 111 or electrospun polymer fibres. 112,113 Multilayer gels have been formed in three dimension, including onion-like structures with multiple layers and internal interfaces. 114,115 Slide ring gels with mobile cross-links have been envisaged. 111 Superporous hydrogels that can quickly imbibe large quantities of water have been developed for drug delivery applications in the gut. 116 Functional gels, such as ‘responsive’ gels 7 , gels that are able to undergo a change in response to a change in local chemical or physical stimulus such as a pH change 5,117 or a change in temperature. 118 Such gels can be used as sensors to detect local environmental changes. 9 Novel properties of gels, such as electroconductivity, 119 may allow for the formation of artificial muscles 9 with good biocompatibility.
Although this review has largely focussed on the limitations of hydrogel stiffness, for demanding applications such as structural tissue engineering, the failure characteristics, strength and toughness, are also critical. There has been significant recent interest in mechanically tough double network gels such as PAAm with poly(2-acrylamido-2-methylpropanesulfonic acid) (PAMPS), for which the toughness of the composite is orders of magnitude greater than the toughness of single component PAAm or PAMPS. 120–122 The mechanisms for extreme toughening have been investigated using molecular modelling. 123
The double network gels in particular show promise for mechanical applications, but as has been demonstrated here, there is a need for a frank discussion within the research community on measurement accuracy and precision when measuring gel mechanical properties. In examination of the papers reviewed here, hardly any mention was found of calibration, or of verification of the testing technique using an independent standard with known physical properties. In general, there is a need for broad consensus to be developed across the field on best practice mechanical characterisation techniques, including both execution of experiments and analysis of data. This is perhaps surprising given the ubiquity of commercially available hydrogels with obvious needs for mechanical stiffness and good transport properties, such as contact lenses. Going forward, in order to develop advanced hydrogel systems for demanding structural tissue engineering applications, and in which the permeability is critical for cell viability, greater confidence in, and even possibly standardisation of, hydrogel mechanical characterisation will be required.
Footnotes
Acknowledgements
The author acknowledges the efforts of DGT Strange and JM Shapiro in reading versions of this manuscript and making helpful corrections and suggestions, and DGTS for drawing Fig. 8b based on an original design of M. Galli, EPFL. The author also acknowledges K Main for hydrogel SEM work in the course of her final year project work (Fig. 4). Finally, Ed Berger, of the University of Virginia, is gratefully acknowledged for first showing the author a plot illustrating the variability of agar modulus with concentration, which inspired this review.