
Abstract
The influence of substitutional alloying elements (Al, Nb and W) on the mechanical properties of MoSi2 with both C11b and C40 structures was explored by first principles calculations. Using the shear modulus/bulk modulus ratio (G/B), Poisson's ratio and Peierls stress, the change in the mechanical properties of the C40 and C11b structured MoSi2 due to the addition of the solute atom has been investigated. The results showed that the ductility of C11b MoSi2 was increased by alloying with the elements (Al and Nb) that form a C40 structured disilicide but decreased by alloying with the elements (W) that form a C11b structured disilicide. In contrast, the effect of alloying elements on the ductility of C40 MoSi2 showed a reverse trend to that of C11b MoSi2.
Introduction
An estimated 10 million people were stranded during last year's eruption of Eyjafjallajökull volcano in Iceland, causing an economic loss of ˜€2bn euros.1 The particles of volcanic ash can bring down an airliner by damaging the thermal barrier coatings used to insulate hot section components of the aircraft engine from high operating temperatures. Therefore, there is presently a strong impetus in the development of novel coating materials that can protect these gas turbine components from volcanic ash damage.2 Of the potential candidates, MoSi2 has received a great deal of attention because of its high melting point (2293 K) and excellent high temperature oxidation and corrosion resistance.3,4 According to the different stacking sequences of close packed planes, molybdenum disilicide can exist in two different forms: a C40 hexagonal structure with ABC stacking sequence along its c axis and a body centred tetragonal C11b structure with {110} atomic planes stacked in an AB type configuration.5 Unfortunately, similar to most high temperature intermetallics, its low toughness at ambient temperature remains a major obstacle to a wide range of practical applications.6 Alloying by chemical substitution for both Mo and Si sites is regarded as an effective strategy to address these deficiencies,7,8 and the effects of alloying with diverse elements on the mechanical properties of both single and polycrystalline MoSi2 have been investigated by both experimental evidence and theoretical calculations.9–12 Among possible substitutional alloying elements, aluminium is apparently most attractive from achieving a unique combination of enhancing the environmental resistance and improving the intrinsic properties of monolithic MoSi2. For instance, a minor substitution of Al for Si in C11b MoSi2 crystal can improve the room temperature ductility, arising from that the addition of Al imparts metallic character to the silicide.13 However, Inui et al.14 had studied the plastic deformation behaviour of C40 Mo(Si1−x,Alx)2 (x = 0·15,0·2) single crystals. They found that the two ternary silicides showed a much higher onset temperature for plastic flow than C11b MoSi2 and hence enhance the brittleness of MoSi2, which was closely associated with the synchroshear mechanism in C40 structured Mo(Si,Al)2.14 However, the effect of alloying with C40 disilicide forming element or C11b disilicide forming element on the mechanical properties of dimorphic MoSi2 crystals is still short of systemic experimental and theoretical evidence so far. In the present study, we have chosen Nb and Al elements that form a disilicide with the C40 structure and the W element that form a disilicide with the C11b structure as alloying element, and investigate the influence of these alloying elements on the mechanical properties of the dimorphic MoSi2 crystals via first principles calculations based on the density functional theory.
Calculation method
The calculations were performed based on the plane wave pseudopotential within the density functional theory using the Cambridge Serial Total Energy Package.15,16 The interactions between the ionic cores and the electrons were defined by the ultrasoft pseudopotentials, and the orbital electrons of Mo-4d55s1, Si-3s23p2, Al-3s23p1, Nb-4d55s1 and W-4d46s2 were treated as valence electrons. The exchange and correlation terms were treated with the Perdew–Burke–Ernzerhof functional in the generalised gradient approximation scheme.17,18 An energy cutoff of 350 eV and a 6×6×4 k point mesh generated by the Monkhorst–Pack method19 were used for all the calculations. The supercells of 3×2×1 and 2×2×1 consisting of 36 atoms for C11b and C40 MoSi2 respectively were constructed and doped with 1–2 W or Nb atom(s), where the corresponding W or Nb concentrations were 0, 0·083 and 0·167 respectively. For alloying C11b MoSi2 by Al, one Si atom is replaced by Al in a C11b supercell (Mo12Si23Al) to maintain its body centred tetragonal (C11b) structure. In the C40 MoSi2 supercell, Si atoms were substituted by one to four Al atom(s), and the corresponding Al concentrations (‘x’) were 0·042, 0·083, 0·125 and 0·167 respectively. The entire structure was allowed to relax until the force on each atom is <0·02 eV Å−1.
Results and discussion
The calculated lattice constants, formation enthalpies (ΔH) and cohesive energies (ΔE) of the binary and ternary MoSi2 with C11b and C40 structures, together with other available experimental data for monolithic MoSi2 with tetragonal and hexagonal structures, are listed in Table 1. The calculated lattice parameters and cohesive energy are in excellent agreement with the values reported by others,20–22 indicating that the computational parameters used are appropriate. All of the binary and ternary MoSi2 alloys show negative values of formation enthalpies and cohesive energies, indicating that these compounds are energetically stable. Moreover, values for both the formation enthalpy and the cohesive energy increase with increasing Al content, suggesting that Al additions lower the thermodynamic stability; however, no discernible trend was noted with variations in W and Nb content.
Calculated properties of binary and ternary MoSi2, including lattice parameters (a and c), formation enthalpies (ΔH) and cohesive energies (ΔE)
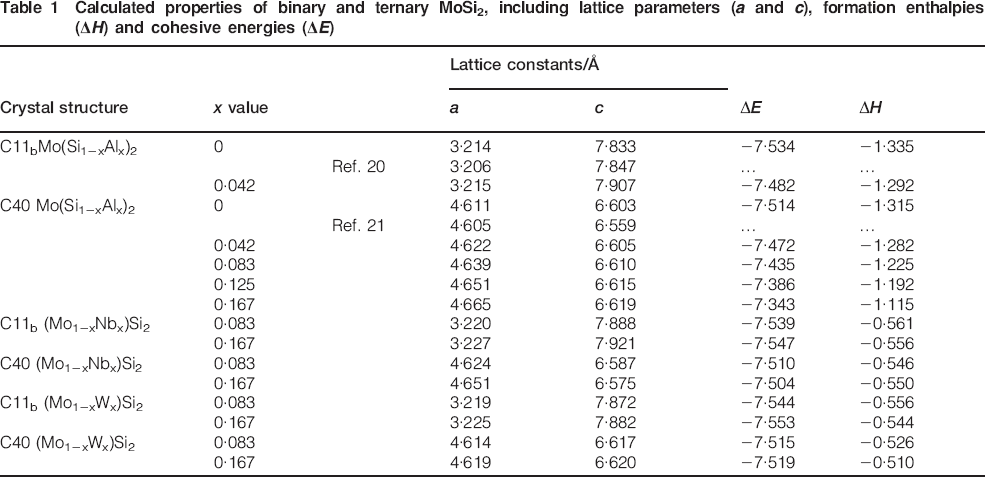
The elastic moduli for binary and ternary MoSi2 with C11b and C40 structures are calculated based on the Voigt–Reuss–Hill approximation,23 and the calculated results are plotted in Fig. 1, along with the available experimental and theoretical values reported in the literature.24,25 As shown in Fig. 1, for C11b structured MoSi2, the calculated shear modulus and bulk modulus deviate from previous experimental data by ∼7 and 1·4; for C40 structured MoSi2, the calculated shear modulus and bulk modulus differ from previously determined values by 1·6 and 3·0. Therefore, with respect to the available data, our calculations yield comparable values. As shown in Fig. 1a, alloying with Nb simultaneously lower the bulk modulus (B) and shear modulus (G) of C11b MoSi2, while for the C40 structure, alloying with Nb reduces the bulk modulus but enhances slightly the shear modulus. The variation of the elastic modulus of MoSi2 with Al addition has a similar tendency with that of Nb addition (Fig. 1b). On the contrary, as C11b disilicide forming element W is added to C11b MoSi2 crystals, bulk modulus (B) is slightly reduced, but shear modulus (G) is increased slightly (Fig. 1c); nevertheless, the opposite trend is observed when W is added to C40 MoSi2 crystals. Generally, the shear modulus is regarded as a good indicator of hardness that is determined by the mobility of dislocations. The shear modulus scales with the stresses required to nucleate or move isolated dislocations and hence is proportional to the hardness of the materials.26,27 As a result, Nb and Al alloying may reduce the hardness of C11b MoSi2, resulting in solid solution softening, and enhance the hardness of C40 MoSi2, resulting in solid solution strengthening, which agrees well with experimental observations by others.28,29 On the contrary, W alloying may increase the hardness of C11b MoSi2 but decrease the hardness of C40 MoSi2, which is consistent with the available experimental data.30
High hardness, when combined well with good ductility, endows a material with excellent toughness. The ratio of bulk/shear modulus G/B has been applied to gauge the ductility of materials.31 That is, if G/B<0·57, the material behaves in a ductile manner; otherwise, the material deforms in a brittle mode. It can be clearly seen from Fig. 1 that the calculated G/B ratios of C11b MoSi2 decrease with increasing addition of Nb and Al, but increase with increasing addition of W, indicating that alloying C11b MoSi2 with Nb and Al is beneficial to the improvement of ductility, and alloying with W is adverse to the ductility. Nevertheless, the opposite trend is observed in C40 MoSi2 alloyed with three kinds of elements. Poisson's ratio can also be used to evaluate the ductility of materials,32 that is, with an increase in Poisson's ratio, ductility is increased accordingly. As seen from Fig. 1, the change of ductility with Nb, Al and W additions evaluated by Poisson's ratio criteria agrees well with that revealed by the G/B ratio.
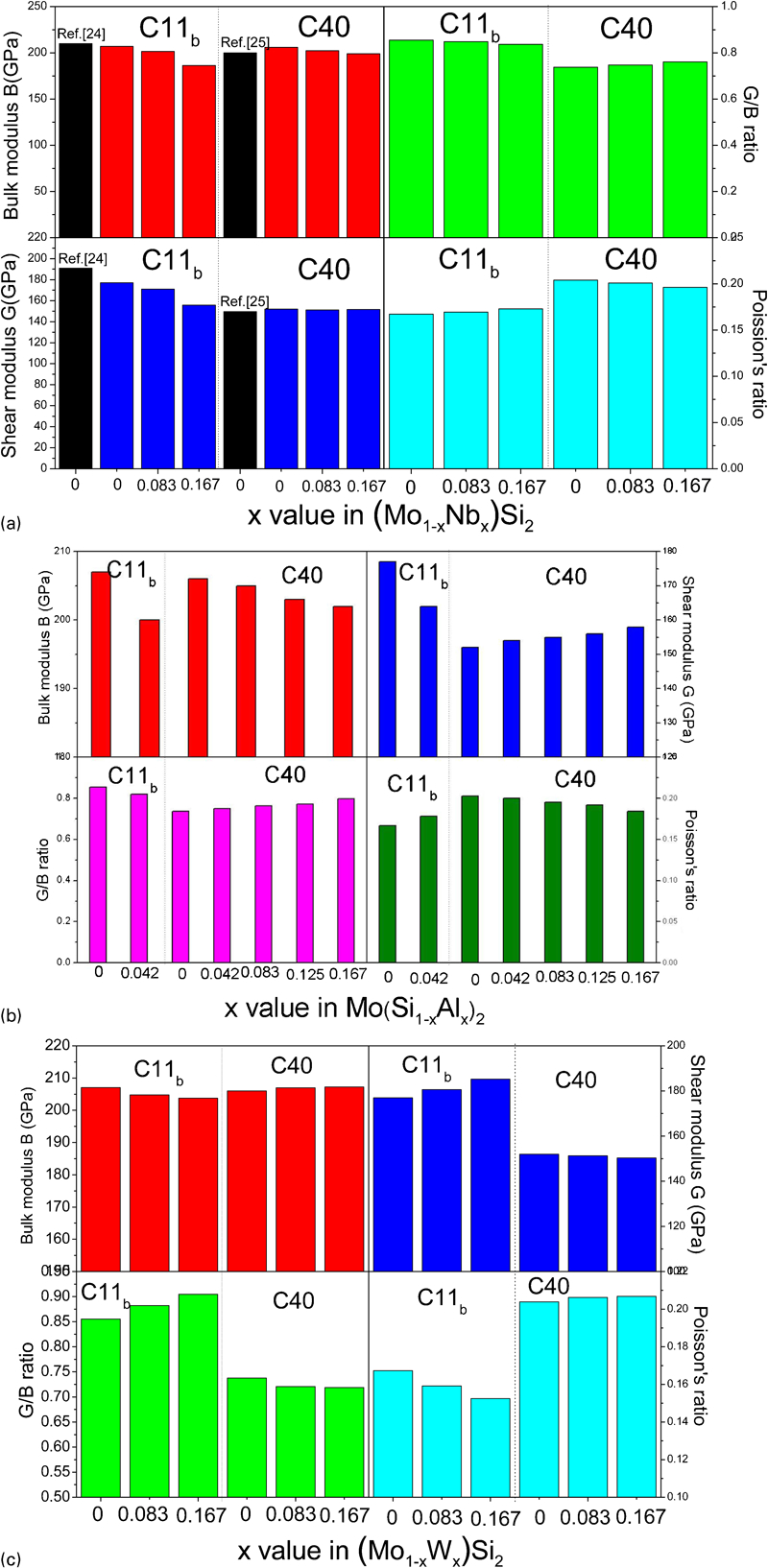
As is well known, the mechanism for plastic deformation involves the generation and movement of dislocations. The mobility of dislocation could be estimated from the Peierls stress (σ) in a glide plane.33,34 Peierls stress can be defined as
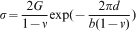
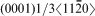 basal slip is active in C40 MoSi2.14 The Peierls stress values calculated from equation (1) are presented in Fig. 2. Because the
basal slip is active in C40 MoSi2.14 The Peierls stress values calculated from equation (1) are presented in Fig. 2. Because the 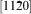 direction in the C40 (0001) plane is equivalent to the [111] direction in the C11b (110) plane, the glide of
direction in the C40 (0001) plane is equivalent to the [111] direction in the C11b (110) plane, the glide of 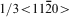 dislocations on {0001} slip planes in the C40 structure corresponds to 1/2<111> glide on {110} planes in the C11b structure.14 An intriguing question arises: why C40 disilicide forming elements Nb and Al and C11b disilicide forming element W have the opposite effects on the Peierls stress for the two crystal structures of MoSi2. This disparity may be explained on the basis of the difference of slip patterns between two slip systems. Because 1/2<111> dislocations of {110}1/2<111> slip system dissociate into two identical 1/4<111> partials separated by a stacking fault.37 For the C11b structured MoSi2, the stacking sequence across the fault is ABC and resembles the stacking of (0001) in the C40 structure. Thus, the addition of C40 disilicide forming element, such as Nb and Al, may reduce the energy difference between C11b and C40 structures, leading to the reduction in energy of the stacking fault and Peierls stress. Conversely, the stacking fault energy and Peierls stress are increased by the addition of C11b disilicide forming element W to C11b MoSi2 crystals. In the case of the C40 structured MoSi2, the
dislocations on {0001} slip planes in the C40 structure corresponds to 1/2<111> glide on {110} planes in the C11b structure.14 An intriguing question arises: why C40 disilicide forming elements Nb and Al and C11b disilicide forming element W have the opposite effects on the Peierls stress for the two crystal structures of MoSi2. This disparity may be explained on the basis of the difference of slip patterns between two slip systems. Because 1/2<111> dislocations of {110}1/2<111> slip system dissociate into two identical 1/4<111> partials separated by a stacking fault.37 For the C11b structured MoSi2, the stacking sequence across the fault is ABC and resembles the stacking of (0001) in the C40 structure. Thus, the addition of C40 disilicide forming element, such as Nb and Al, may reduce the energy difference between C11b and C40 structures, leading to the reduction in energy of the stacking fault and Peierls stress. Conversely, the stacking fault energy and Peierls stress are increased by the addition of C11b disilicide forming element W to C11b MoSi2 crystals. In the case of the C40 structured MoSi2, the 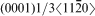 basal slip appears to occur through a synchroshear mechanism in which a sequence of shears occurs synchronously on two adjacent (0001) planes, which may induce a C40→C11b phase transformation.14 Therefore, with the increasing amount of Nb and Al addition, the energy difference between C40 and C11b structures increases steadily, and the Peierls stress changes accordingly. Instead, with the increasing addition of W element that forms a disilicide with the C11b structure, the energy difference between C40 and C11b structures decreases gradually, and thus, the reverse trend is obseved for Peierls stress.
basal slip appears to occur through a synchroshear mechanism in which a sequence of shears occurs synchronously on two adjacent (0001) planes, which may induce a C40→C11b phase transformation.14 Therefore, with the increasing amount of Nb and Al addition, the energy difference between C40 and C11b structures increases steadily, and the Peierls stress changes accordingly. Instead, with the increasing addition of W element that forms a disilicide with the C11b structure, the energy difference between C40 and C11b structures decreases gradually, and thus, the reverse trend is obseved for Peierls stress.
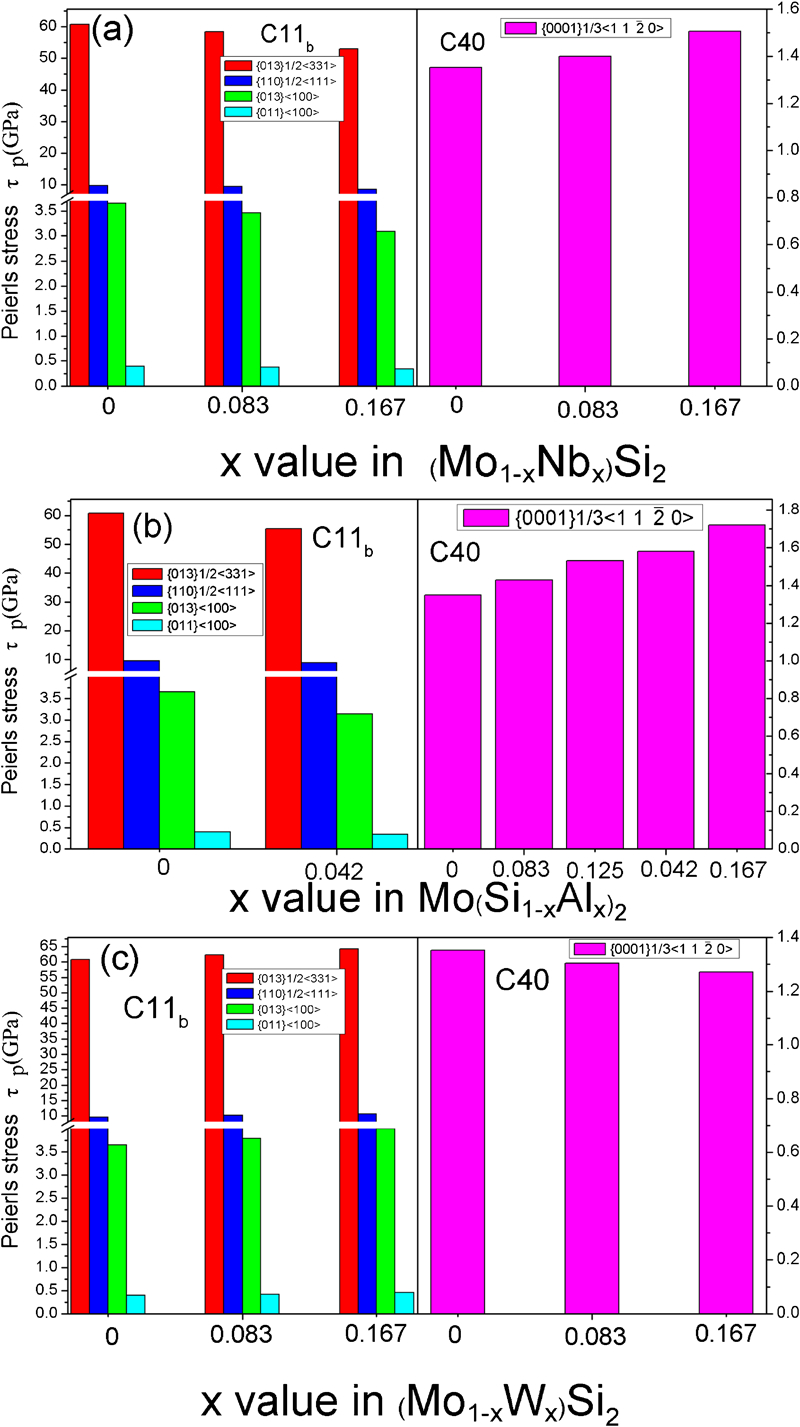
Calculated Peierls stress (τp) for C11b and C40 structured a (Mo1−xNbx)Si2, b Mo(Si1−xAlx)2 and c (Mo1−xWx)Si2
On the basis of the above calculated results, it can be concluded that the ductility of C11b MoSi2 is increased by alloying with elements that form a C40 disilicide and is decreased by alloying with elements that form a C11b disilicide, whereas the effect of the alloying element on the ductility of C40 MoSi2 is running counter to that found in C11b MoSi2. To develop a better understanding of the effect of Nb, Al and W alloying elements on the ductility of MoSi2, the electronic structures of the binary and ternary MoSi2 with C11b and C40 crystals are investigated. Figure 3a–d shows the density of states (DOS) of the binary and ternary MoSi2 with C11b structures. The most evident feature for these alloys is their metallic nature according to their Fermi level state. It is clear that the value of the DOS at Fermi level increases with the increase in Al and Nb alloying but decreases by W alloying. This result suggests that alloying with Nb and Al may introduce a more metallic nature to the bonding, resulting in the enhancement in ductility of C11b MoSi2; on the contrary, W alloying increases the covalent nature of the bonding, causing the low ductility of C11b MoSi2. This is consistent with the above analysis for the G/B ratio and Peierls stress. Furthermore, there is a deep valley close to the Fermi level (EF), and this valley is referred to as a pseudogap. The presence of a pseudogap indicates directionality of bonding in the C11b structure. Directional bonding can effectively resist elastic and plastic deformations and contribute to high elastic moduli and strengths,38 which is responsible for the high G/B values of the C11b structure. On the left of the pseudogap, there exists strong hybridisation of Mo-d, Mo-p and Si-p orbitals between −3 and −1 eV below the Fermi level (EF) in C11b MoSi2 (Fig. 3a). However, when alloying elements are added to C11b MoSi2 (Fig. 3b–d), the Si-p states exhibit a weak interaction with the Nb-d and Al-p states from −3 to −1 eV and show a much stronger interaction with the W-d states in this area. This finding answers the question why C40 disilicide former element (Nb and Al) and C11b disilicide former element (W) have the opposite influences on the ductility of C11b MoSi2. The DOSs of the binary and ternary MoSi2 alloys with C40 structures are shown in Fig. 3e–h respectively. The DOS of C40 MoSi2 near the Fermi level (EF) is similar to that of C11b MoSi2. The strength of σ bond of Si–Si (strong hybridisation of Si-s states with Si-p states) located between −14 and −7 eV in C40 MoSi2 is lower than that of the corresponding σ bond in C11b MoSi2. Moreover, the intensity of the strong hybridisation peak of Mo-d, Mo-p and Si-p orbitals between −3 and −1 eV below the Fermi level (EF) in C40 MoSi2 is less than that in C11b MoSi2 structures. The results indicate that the covalent bond strength in C40 MoSi2 structure is weaker than that of the C11b MoSi2 structure, indicating that the ductility of C40 MoSi2 is superior to that of C11b MoSi2. On the other hand, the DOS of the ternary C40 (Mo1−xNbx)Si2, Mo(Si1−xAlx)2 and (Mo1−xWx)Si2, on the whole, is remarkably similar to that of C40 MoSi2. In addition, after substituting Mo with Nb, Al or W elements, the σ bonds remain quite stable, while the hybridisation of Mo-d, Mo-p and Si-p orbitals between −3 and −1 eV below the Fermi level (EF) undergoes a noticeable change in the position and height of the peak. This hybridisation state between −3 and −1 eV shifts upward, together with a slight decrease in peak height by Nb alloying, but this hybridisation state evidently shifts downward by W alloying. This result in C40 disilicide forming element and C11b disilicide forming element has the opposite effect on the mechanical properties of C40 MoSi2.
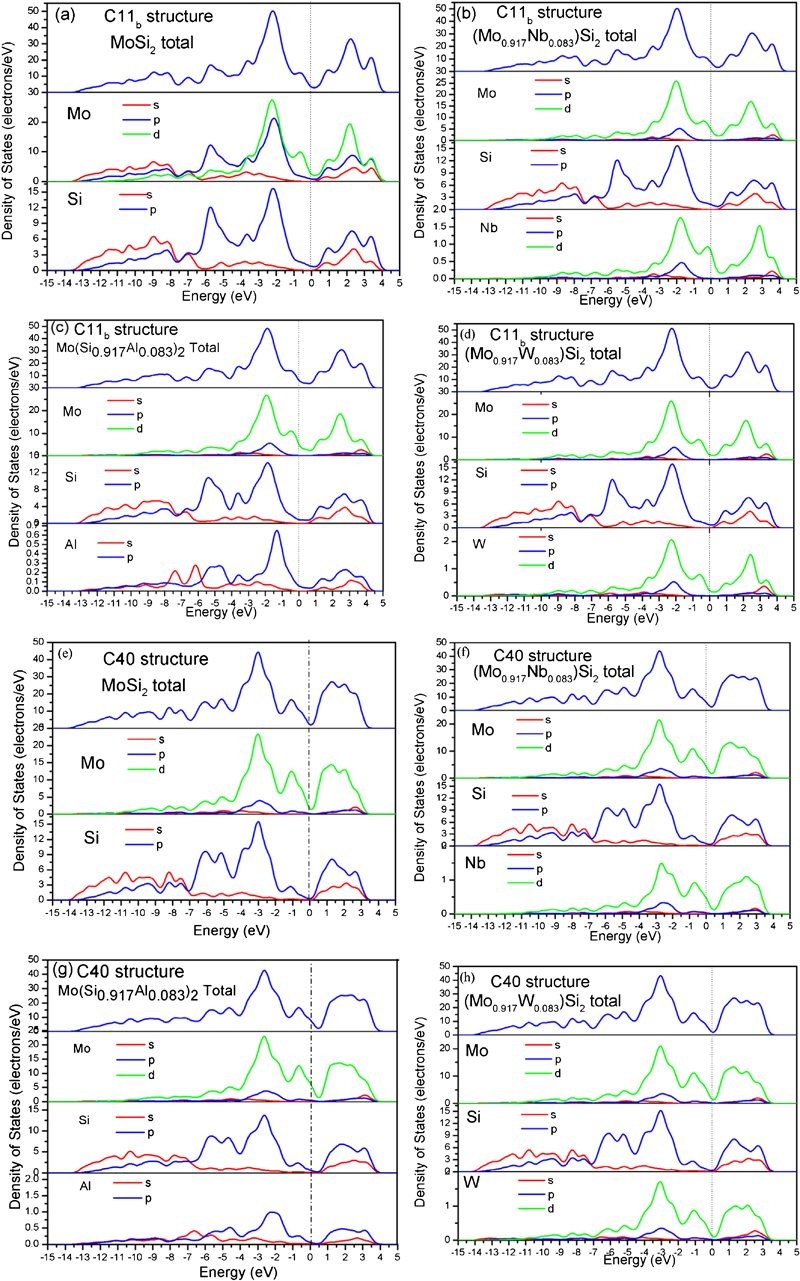
Total and partial DOS of a C11b MoSi2, b C11b (Mo0·917Nb0·083)Si2, c C11b Mo(Si0·917Al0·083)2, d C11b (Mo0·917W0·083)Si2, e C40 MoSi2, f C40 (Mo0·917Nb0·083)Si2, g C40 Mo(Si0·917Al0·083)2 and h C40 (Mo0·917W0·083)Si2: dot lines represent position of Fermi level (EF)
To further reveal the nature of bonding in these materials, the charge density difference is studied and plotted in Fig. 4. The (110) electron charge distribution of C11b MoSi2 is shown in Fig. 4a. Strong charge correction regions appear around the Mo and Si atoms, confirming the existence of covalent bonding between Mo and Si. This strong covalent bonding makes dislocation motion difficult and leads to a high Peierls stress. Moreover, the Si–Si bonds along the [001] direction show an ionic–covalent nature, arousing the absence of slip on {001} and low cleavage energies for the [001] direction.15,39 In the case of C11b Mo(Si0·958Al0·042)2 (Fig. 4b), the changes in charge densities are localised near the Al site, and the strength of the Si–Si bond along the [001] direction slightly weakens. For the binary C40 MoSi2 (Fig. 4c), the directional bonding between Mo and Si atoms is clearly observed, but the strength of the Mo–Si bonds is weaker than that of C11b MoSi2. For the ternary C40 MoSi2 (see Fig. 4d), the charge accumulation between the Si–Al atoms is stronger than that between the Si–Si atoms, which explains why C40 MoSi2 becomes more brittle with an increase in Al content.
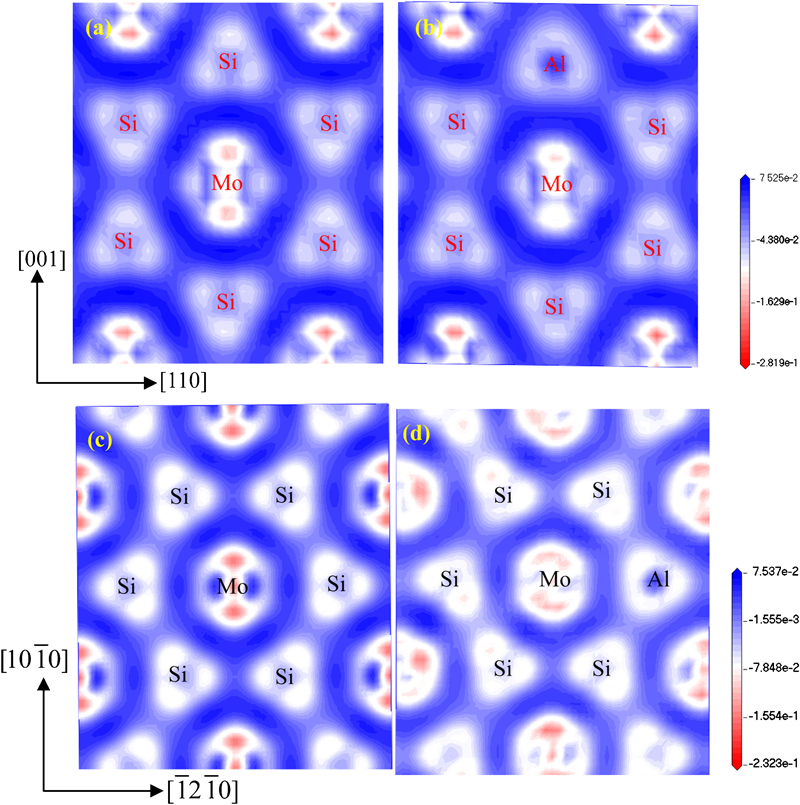
Charge density difference on (110) plane of a C11b MoSi2 and b C11b Mo(Si0·958Al0·042)2 and (0001) plane of c C40 MoSi2 and d C40 Mo(Si0·958Al0·042)2: positive (negative) values denote charge accumulation (depletion)
Conclusions
In summary, the influence of Nb and Al elements that form a disilicide with the C40 structure and that of W element that forms a disilicide with the C11b structure on the mechanical properties of MoSi2 with both C11b and C40 structures are investigated under first principles calculations. The calculated results show that alloying with Nb and Al simultaneously lowers the bulk modulus (B) and the shear modulus (G) of C11b MoSi2, while for the C40 structure, alloying with Nb and Al reduces the bulk modulus but enhances slightly the shear modulus. On the contrary, as C11b disilicide forming element W is added to C11b MoSi2 crystals, the bulk modulus (B) is slightly reduced, but the shear modulus (G) is increased slightly; nevertheless, the opposite trend is observed when W is added to C40 MoSi2 crystals. The change of ductility of the C40 and C11b structured MoSi2 with alloying elements is explained according to G/B ratio, Poisson's ratio and Peierls stress. The results show that the ductility of C11b MoSi2 increases by alloying with C40 disilicide forming element and decreases on alloying with C11b disilicide forming element, but the ductility of C40 MoSi2 increases by alloying with C40 disilicide forming element and increases on alloying with C11b disilicide forming element. The electronic structure analysis indicates that alloying with Nb and Al introduces a more metallic nature to the bonding, resulting in the enhancement in ductility of C11b MoSi2; on the contrary, W alloying increases the covalent nature of the bonding, causing the low ductility of C11b MoSi2.
Footnotes
Acknowledgements
The authors acknowledge the financial support of the National Natural Science Foundation of China under grant no. 51175245 and the Key Program of Jiangsu province Natural Science Foundation of China under project no. BK2010073. Z. H. Xie and P. Munroe also acknowledge the financial support provided by Edith Cowan University (ECU) through the ECU-Industry Collaborative Scheme.