
Abstract
Many technologically important materials exhibit paramagnetism, for example, lithium intercalation materials for lithium-ion batteries and platinum nanoparticles for catalysis. In order to study their properties, for example, their electron transport properties or catalytic efficiency, and hence tailor them to specific engineering applications, information on their structure and bonding needs to be acquired. Solid state nuclear magnetic resonance (NMR) is an excellent experimental method for achieving this; however, the resulting spectra are often difficult to interpret. Calculations based on quantum mechanics therefore provide an invaluable aid for decoding these experimental spectra.
The dominant interaction in paramagnetic materials is the hyperfine interaction. This is the interaction between the spin of a nucleus and the spin of an unpaired electron. The hyperfine interaction affects the appearance of NMR spectra, both by shifting the frequencies at which resonance occurs and by removing degeneracies so that a single resonance frequency splits into several frequencies. This review examines the methods and current limitations for calculating hyperfine interaction parameters, placing this in the context of experimental NMR investigations for lithium intercalation materials and platinum nanoparticles. In this way, the importance of developing increasingly accurate methods for calculating hyperfine parameters is illustrated.
Literature review motivation
Nuclear magnetic resonance (NMR) is an extremely powerful and flexible experimental technique. It employs the sensitivity of a nuclear spin to its atomic environment to probe the structure and bonding of the material and hence provide insight into the material's mechanical, electrical, magnetic and chemical properties. Owing to its high sensitivity and transferability, NMR now forms part of the standard tool kit at the experimentalist's disposal.
The large number of interactions influencing the behaviour of the nuclear spins results in experimental spectra being difficult to analyse. While it is often possible to interpret experimental NMR spectra for liquids, solid-state spectra present a greater challenge. This is because the inherent anisotropy of solids leads to a high degree of broadening of the peaks about the resonant frequencies, which makes the individual contributions of different interactions harder to separate. Simulated NMR spectra are therefore an invaluable tool for decoding the experimental spectra. They can also be used to predict the properties of exotic materials in advance of conducting experiments and therefore guide the experiments themselves.
The simulations for solid-state NMR are obtained using a first-principles approach, without the use of empirical parameters. Since the resonance of nuclear spins is a fundamentally quantum mechanical phenomenon, the calculations are based on solving the quantum mechanics of the system. Such calculations would not have been possible in practice were it not for theoretical advances such as density functional theory (DFT), the development of iterative algorithms as implemented in modern codes and high performance computers.
It is currently possible to calculate relevant NMR parameters for diamagnetic materials.1–3 These calculations have proved so successful that the methodology has been widely adopted by the experimental community.4,5 The methodology for first-principles calculations of NMR parameters for paramagnetic materials (those materials containing unpaired electrons) has yet to be fully developed.
The dominant interaction in the NMR of paramagnetic materials is the hyperfine interaction. This is the interaction between the spin of a nucleus and the spin of an unpaired electron. Of particular importance is the isotropic component of the hyperfine interaction, the Fermi contact term, which manifests in a wide range of materials science applications, including lithium-ion cathode materials and platinum nanoparticles for catalysis.
Introduction to magnetic resonance
In 1922, Stern and Gerlach discovered that nuclei possess their own intrinsic magnetic moment (spin). 6 Just over a decade later, Stern, continuing his molecular beam experiments along with Estermann and Frisch, succeeded in measuring the magnetic moment of the proton.7–10
Rabi is often credited with inventing the magnetic resonance technique. In 1938, building on Stern and other's achievements and the under-acknowledged work of Gorter, 11 he and his group succeeded in measuring the frequency at which the magnetic moment of lithium atoms resonated in a beam of LiCl molecules (see Fig. 1. 12 A year later, they measured the magnetic resonances of Li6, Li7 and F19 in LiCl, LiF, Li2 and NaF molecules. 13 His achievements earned him the 1944 Nobel Prize in Physics. 14
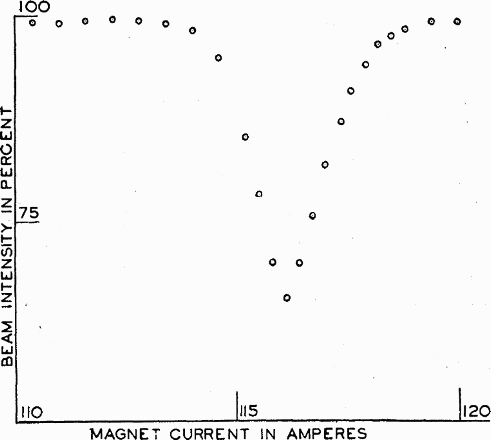
The first magnetic resonance spectrum from Rabi et al. 12 (reproduced in Hurwitz 16 ). This was obtained by Rabi et al. for lithium in a molecular beam of LiCl. ‘MAGNET CURRENT IN AMPERES’ relates to the magnitude of the homogeneous magnetic field. Rabi and his group found that this produced more accurate results than the modern method of keeping the magnetic field constant and varying the frequency of the radiation. ‘BEAM INTENSITY IN PERCENT’ refers to the intensity of the LiCl beam. The plot minimum occurs at the resonance frequency of the lithium atoms (reprinted from Ref. 12 with permission: © 1938 American Physical Society)
Since then, the supreme sensitivity and transferability of the technique has resulted in countless applications in chemistry, physics, biology, materials science and medical imaging, and has produced an additional six Nobel Prizes. An excellent review on the early history of magnetic resonance has been written by Ramsey. 15
Principle of magnetic resonance
Nuclei possess an intrinsic spin angular momentum, 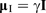
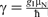 , where gI is the dimensionless g-factor for a free nucleus, μN is the nuclear magneton and ħ is the reduced Planck constant. The magnitude of γ is specific to each isotope and is expressed in terms of 107 rad T− 1 s− 1, or sometimes MHz− 1 T− 1, with a conversion factor of 2π between the two units.
, where gI is the dimensionless g-factor for a free nucleus, μN is the nuclear magneton and ħ is the reduced Planck constant. The magnitude of γ is specific to each isotope and is expressed in terms of 107 rad T− 1 s− 1, or sometimes MHz− 1 T− 1, with a conversion factor of 2π between the two units.
When there is no external magnetic field, all nuclear spin
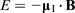
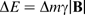
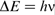 (where h is Planck's constant) and the transition selection rule, Δm = ± 1, are satisfied. Thus, the frequency of the radiation required for NMR, termed the Larmor frequency, is
(where h is Planck's constant) and the transition selection rule, Δm = ± 1, are satisfied. Thus, the frequency of the radiation required for NMR, termed the Larmor frequency, is
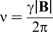
The additional local magnetic field, 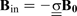
Experimentally, the chemical shift, δ, is measured relative to a standard reference σref so that δ is conventionally defined as
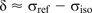
Hyperfine interaction
In the field of magnetic resonance, the hyperfine interaction is defined as the interaction between the spin of the nucleus,
The Hamiltonian for hyperfine coupling, 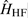 , can be expressed as the interaction between the magnetic dipole moment,
, can be expressed as the interaction between the magnetic dipole moment, 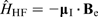
Since the nuclear spin magnetic dipole moment, 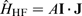
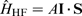
Incorporating into the hyperfine Hamiltonian the interaction between the electron and M nuclear spins 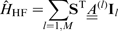
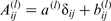
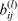 is the anisotropic hyperfine (spin-dipolar) interaction, δ
ij
is the Kronecker delta, and i and j are cartesian axes.
is the anisotropic hyperfine (spin-dipolar) interaction, δ
ij
is the Kronecker delta, and i and j are cartesian axes.
Physically the Fermi contact interaction is the magnetic (spin–spin) interaction between the electron and the nucleus when the electron is inside the nucleus. The strength of the Fermi contact interaction is directly proportional to 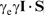 (Ref. 23, p. 465) and can therefore be expressed in atomic units as
(Ref. 23, p. 465) and can therefore be expressed in atomic units as
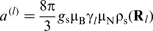
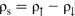 ) at the centre of the lth nucleus, γ
l
is the gyromagnetic ratio for the lth nucleus, gs is the dimensionless g-factor for a free electron, γe is the gyromagnetic ratio for a free electron, μB is the Bohr magneton and μN is the nuclear magneton.
22
Since ρs is directly proportional to the square of the electron wavefunction at the centre of the nucleus,
) at the centre of the lth nucleus, γ
l
is the gyromagnetic ratio for the lth nucleus, gs is the dimensionless g-factor for a free electron, γe is the gyromagnetic ratio for a free electron, μB is the Bohr magneton and μN is the nuclear magneton.
22
Since ρs is directly proportional to the square of the electron wavefunction at the centre of the nucleus,  , the Fermi contact interaction is only affected by s-like electrons, because only s-like wavefunctions are non-zero at the nucleus. The calculation of
, the Fermi contact interaction is only affected by s-like electrons, because only s-like wavefunctions are non-zero at the nucleus. The calculation of 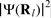 poses several computational problems outlined in the ‘First-principles calculations of hyperfine parameters’ section. By contrast, the spin–dipolar interaction is affected by non s-like electrons.
poses several computational problems outlined in the ‘First-principles calculations of hyperfine parameters’ section. By contrast, the spin–dipolar interaction is affected by non s-like electrons.
The hyperfine interaction emerges as a large measurable shift in resonance frequencies in paramagnetic NMR spectra. It is related to the Knight shift in the NMR of metallic systems (see the next section).
Knight shift
First observed by Knight in 1949,
24
this is a systematic shift in the resonance frequencies of metallic systems, which is not observed in the resonance frequencies of the same element in non-metallic systems. To a first approximation, this shift occurs because an external magnetic field will induce a magnetisation, M, in the paramagnetic material. The magnetisation arises from the unequal change in energy of the conduction electrons (those close to the Fermi energy, EF) between those with spin- and those with spin-
and those with spin-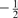 . Thus, the material acquires a magnetisation, characterised by the average spin of the conduction electrons ⟨Sz⟩ or by the Pauli spin susceptibility χs, where
. Thus, the material acquires a magnetisation, characterised by the average spin of the conduction electrons ⟨Sz⟩ or by the Pauli spin susceptibility χs, where 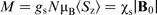 (where gs is the free electron g-factor and N is Avogadro's number). This net electron spin creates a magnetic field, additional to
(where gs is the free electron g-factor and N is Avogadro's number). This net electron spin creates a magnetic field, additional to 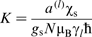
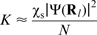
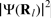 poses some difficulties as outlined in the ‘First-principles calculations of hyperfine parameters’ section.
poses some difficulties as outlined in the ‘First-principles calculations of hyperfine parameters’ section.
The magnitude of the Knight shift at each crystallographically distinct nucleus in a sample will differ. This is observed as an inhomogeneous broadening in the resonance peak. 26
Solid-state NMR
The main difficulty when acquiring experimental solid-state NMR spectra is that there are very large amounts of line broadening due to the inherent anisotropy of the solid samples. This leads to difficulties pertaining to the sensitivity and resolution of spectra. Notable anisotropic interactions include shielding/chemical shift anisotropy (CSA), dipolar coupling (the interaction between two magnetic moments through space (Ref. 18, p. 24)) and quadrupolar coupling (an electric interaction affecting nuclei with spin greater than  (Ref. 18, p. 28)). While it may be desirable to remove these interactions from spectra to aid interpretation, the interactions themselves can provide further valuable information about the sample. Dipolar coupling provides information on the distances between nuclei and atomic structure, CSA can provide structural information and J-coupling can provide information on chemical bonding and geometry. There therefore exist methods for selectively reintroducing interactions in spectra. An overview of the methods to both remove and reintroduce interactions is presented in Bonhomme et al.'s extensive review paper.
4
(Ref. 18, p. 28)). While it may be desirable to remove these interactions from spectra to aid interpretation, the interactions themselves can provide further valuable information about the sample. Dipolar coupling provides information on the distances between nuclei and atomic structure, CSA can provide structural information and J-coupling can provide information on chemical bonding and geometry. There therefore exist methods for selectively reintroducing interactions in spectra. An overview of the methods to both remove and reintroduce interactions is presented in Bonhomme et al.'s extensive review paper.
4
The most common technique for removing broadening from CSA and dipolar coupling is MAS. Relevant interactions in NMR involve a dependency on the orientation of the crystal relative to the applied magnetic field of the form 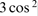 θ − 1, where θ is the angle between the crystallographic principal axis and the applied magnetic field. Associated broadening can therefore be removed by rapidly spinning the sample at the ‘magic angle’ 54.736° to the applied magnetic field. A detailed explanation can be found in Harris et al.'s book on solid-state NMR (Ref. 18, p. 32). Rotation rates tend to be 5–70 kHz
4
; however, rates of up to 110 kHz may be used.
27
In the case of paramagnetic materials, where there are unpaired electrons present, the anisotropy associated with the hyperfine interaction can be very large and therefore very high rotation rates are required in order to achieve spectra of a suitable resolution (Ref. 18, p. 213).
θ − 1, where θ is the angle between the crystallographic principal axis and the applied magnetic field. Associated broadening can therefore be removed by rapidly spinning the sample at the ‘magic angle’ 54.736° to the applied magnetic field. A detailed explanation can be found in Harris et al.'s book on solid-state NMR (Ref. 18, p. 32). Rotation rates tend to be 5–70 kHz
4
; however, rates of up to 110 kHz may be used.
27
In the case of paramagnetic materials, where there are unpaired electrons present, the anisotropy associated with the hyperfine interaction can be very large and therefore very high rotation rates are required in order to achieve spectra of a suitable resolution (Ref. 18, p. 213).
Despite the plethora of different techniques available for probing the structure, interactions and chemical environments in solids, the experiments are time consuming and restricted to certain systems, and it can be difficult to acquire spectra of sufficient resolution for analysis. Theoretical calculations of relevant NMR parameters therefore provide a much needed, if not essential, aid for the prediction and interpretation of experimental spectra in addition to valuable insights into the active mechanisms in a system.
Introduction to first-principles calculations
First-principles approaches aim to calculate properties of materials using only the postulates of quantum mechanics. 28 Since the mechanisms involved in NMR are quantum mechanical in origin, calculating their relevant parameters through this approach is most appropriate.
For a system containing electrons and nuclei, the time independent nonrelativistic Schrödinger equation, 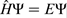 , can be expressed as
, can be expressed as
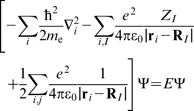
Density functional theory
One method for simplifying equation (15) is the Hartree–Fock method. This involves treating the interaction between electrons as an interaction between a single electron and the mean field of all the other electrons. 29 However, this is still computationally expensive for solid systems. DFT is a significantly cheaper alternative and has been widely adopted by theoretical and experimental communities for calculating NMR parameters. 4
The DFT formalism begins from the premise that the total ground state energy, E0, of a system is a unique functional of the total electron density, ρ, of the system, as formally proved by Hohenberg and Kohn in 1964
30
:
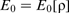
Thus, equation (15) becomes a set of single electron equations, which are coupled together through the electron charge density ρ(
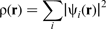
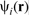 is the single electron wavefunction.
is the single electron wavefunction.
The total energy E of the system can therefore be expressed as

These equations can be solved iteratively (see Fig. 2). However, to begin with, Exc[ρ] still has to be approximated.
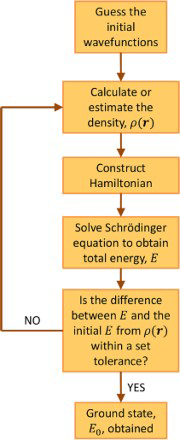
The iterative solving of the Kohn–Sham equations for the total energy, E. Self-consistency is reached when the difference in E between a set number of successive iterations is within a set tolerance
Exchange–correlation functionals
The simplest approximation for Exc[ρ] is the local density approximation (LDA). This assumes that Exc[ρ] depends only on the charge density at each point.32,33 By additionally taking into account the gradient at each point, more advanced generalised gradient approximation (GGA) exchange–correlation functionals are obtained. The most commonly used exchange–correlation functional used for calculating NMR parameters is the GGA functional formulated by Perdew, Burke and Ernzerhof (PBE). 34
Practical implementation of DFT
In order to solve equation (18) in practice, a set of numerical approximations need to be made.
Basis set
The wavefunction needs to be expressed as a ‘linear combination of simple mathematical functions’.
4
These could be set of localised atom-centred orbitals, for example, the Gaussian type orbitals used in the CRYSTAL
35
and SIESTA
36
codes. Another possibility is a set of planewaves as used in the CASTEP code
37
:
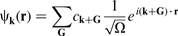
Periodicity of structures
In order to reduce the number of atoms in the solid-state system to a computationally realistic level, the periodicity of the crystal lattice is exploited using Bloch's theorem (Ref. 25, p. 163):
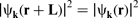
k point sampling
Calculating the charge density at a set of discrete points in the Brillouin zone provides a sufficiently accurate sum of the total charge density if enough points are sampled,
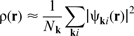
Pseudopotentials
Valence wavefunctions oscillate rapidly near the nucleus and therefore require a high energy cutoff in order to be accurately described. To reduce the energy cutoff, and thus increase computational efficiency, the potential close to the nucleus, defined within a radius r c , is smoothed. This results in a pseudopotential and a correspondingly smoothed pseudowavefunction. Crucially, the energy eigenvalues are not affected. This is the pseudopotential approximation.
In most cases, only the valence electrons require pseudopotentials. The eigenvalues for the core electrons, since they are not involved in bonding, are assumed to be the same as those in an isolated atom and can therefore be easily calculated. This is the frozen core approximation. In the case of hyperfine parameters, this is not a valid approximation.
First-principles calculations of hyperfine parameters
The initial theory for calculating NMR parameters was developed by Ramsey in the 1950s.42–44 Since then, computational methods pertaining to the NMR parameters for diamagnetic materials have been developed including the magnetic shielding tensor, σ, as defined in equation (5).1,2 For metallic systems, there has been progress in the calculation of magnetic shielding tensors and Knight shifts. 41 In terms of hyperfine parameters, early qualitative attempts to calculate Fermi contact shifts involved calculations on molecular solids using Hartree–Fock and DFT.45–48 There is also wide interest in calculating the hyperfine interaction for single molecules (Refs. 29, 49, 50 and 51, p. 209).
Methodologies and challenges
In order to quantitatively calculate hyperfine parameters within the methodology outlined in the ‘Density functional theory’ section, two challenges must be overcome:
Valence electrons—the wavefunctions for these are smoothed pseudowavefunctions within r
c
. This leads to non-physical charge densities close to the nucleus and thus incorrect hyperfine parameters. Core electrons—these are calculated using the frozen core approximation. However, the spin of the valence electrons can cause spin polarisation of the core electrons. Because accurately computing the Fermi contact interaction relies on exactly knowing the spin density (
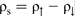 ) at the nucleus, failure to include this core polarisation will lead to significant errors.
) at the nucleus, failure to include this core polarisation will lead to significant errors.
Projector augmented wave method
Blöchl overcame the first challenge by recreating the true all-electron non-pseudised wavefunction for valence electrons from the pseudowavefunction within a region close to the nucleus bounded by a selected radius, rc. 52
The pseudowavefunction  is mapped onto the all-electron wavefunction
is mapped onto the all-electron wavefunction  by the operator
by the operator  according to
according to
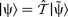
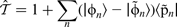
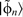 . Equation (23) therefore corresponds to removing the section of
. Equation (23) therefore corresponds to removing the section of 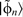 located within r
c
and replacing it with the equivalent all-electron atomic states
located within r
c
and replacing it with the equivalent all-electron atomic states 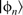 .
. 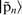 is a function strongly localised within rc and is therefore used to ensure that only the section of
is a function strongly localised within rc and is therefore used to ensure that only the section of 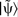 within rc is altered. A pictorial representation of equation (23) is given in Fig. 3.
within rc is altered. A pictorial representation of equation (23) is given in Fig. 3.
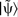
Schematic of the projector augmented wave (PAW) method.
53
The x axis signifies the radial distance from the nucleus: a initial pseudowavefunction
; b two separate pseudo-atomic states 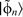 ; c two corresponding all-electron atomic states
; c two corresponding all-electron atomic states 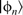 ; d final reconstructed all-electron wavefunction
; d final reconstructed all-electron wavefunction 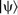 (reprinted from Ref. 53 with permission: © 2010 Wiley)
(reprinted from Ref. 53 with permission: © 2010 Wiley)
Extending the PAW methodology to the Fermi contact interaction results in the associated operator
53
:
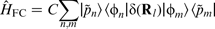
Core polarisation
Progress in overcoming the second challenge was begun by Yazyev et al. through the core spin–polarisation correction method. 54 Instead of calculating the core electron eigenstates from an isolated atom, the all-electron wavefunction for the core electrons was reconstructed in the presence of the valence electrons, with the valence states being approximated to those in an isolated atom. This produced a significant improvement on the Fermi contact term for a series of small molecules. Declerck et al. proposed an alternative hybrid method for the inclusion of core polarisation, which combined an all-electron calculation for the atom of interest with a pseudopotential calculation for all the others. 55 Not only could this hybrid method be applied to single atoms and small molecules, but it could also be applied to solids. The hyperfine interaction of the L-α-alanine R2 radical in a crystal of L-α-alanine molecules was calculated as an example of this. Bahramy et al. developed a further improvement through calculating the core polarisation using first-order perturbation theory.56,57 In this way, the spin density at the nucleus was evaluated in two steps. First, the PAW method of Blöchl was used to reconstruct the all electron wavefunctions of the valence electrons within r c . Second, the contribution to core polarisation from the core electrons was evaluated using first-order perturbation theory. 57 This methodology has been used by Filidou et al. 22 to evaluate Fermi contact terms for small molecules and fullerene derivatives.
Materials applications
Lithium-ion battery materials and platinum nanoparticles are two paramagnetic systems that exhibit hyperfine interactions and are of significant importance to current and future engineering applications. The first-principles modelling methods discussed in the ‘Introduction to first-principles calculations’ and ‘First-principles calculations of hyperfine parameters’ sections provide an invaluable aid in the determination of their materials properties.
Lithium-ion battery materials
Lithium intercalation materials, as discussed here, are layered oxides with lithium ions in between the layers. Figure 4 shows the compound LiCoO2 which formed the cathode in the first commercial Li-ion battery. 58 This has been widely used in laptops, mobile phones and cameras; however, the low abundance of cobalt makes it expensive to manufacture and it has a low theoretical charge capacity of ∼130 mAh g− 1. 58 As a result, research on other lithium compounds is vast. 58
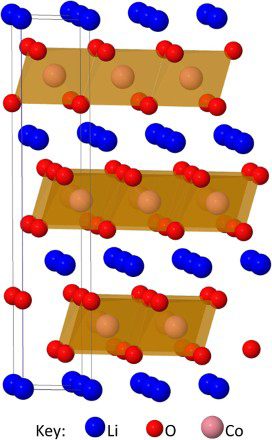
Structure of LiCoO2, showing the layers of CoO6 octahedra and Li+ ions
Solid-state NMR studies of cathode materials for lithium-ion batteries are extensive.59,60 Spectra are dominated by interactions between electronic and nuclear spins, and exhibit Knight shifts (between the conduction electrons and nuclei) and other hyperfine shifts (between unpaired electrons and nuclei). Often, the 7Li and 6Li resonances are studied.61–65 This is because neighbouring paramagnetic metal ions induce an electron spin density at the lithium nucleus, which experimentally emerges as large hyperfine shifts. Analysis of these hyperfine shifts provides insight into conduction mechanisms and structural changes from electrochemical recycling.
The hyperfine shift can be understood to originate from two competing mechanisms: spin delocalisation (or hybridisation) and spin polarisation.
Experimental hyperfine shifts
The Fermi contact shift has been experimentally measured in LiCoO2, 66 and the related structure LiMnO2. 67 These have an ABCABC layered ‘O3’ structure as characterised by Delmas et al. in their structural classification of the layered oxides. 68 Marichal et al. studied nickel doping of LiCoO2. 61 Other candidate compounds for which the hyperfine shifts have been measured include chromium doped LiCoO2 66 ; the spinels LiMn2 − x M x O4, for which LiMn2O4 has a large hyperfine shift >500 ppm in 6Li and 7Li MAS spectra69–72; and lithium phosphates LiMPO4, where M is Mn, Fe, Ni, Co,73,74 La4LiNiO8, 75 La4LiMnO8, 76 and La3SrLiMnO8. 76
First-principles calculations
The first calculations of the hyperfine interaction in lithium-ion battery materials were conducted by Carlier et al. 48 They used DFT (see the ‘Density functional theory’ section) to calculate the electron spin density of the system and then integrated this within the vicinity of the nucleus to obtain the total electron spin density contributing to the Fermi contact shift. They used an integration radius of 0.8 Å, compared to the measured radius of a lithium ion in an octahedral site of 0.76 Å. 77 It is important to note that they used pseudopotentials with no PAW correction, which meant that their calculated charge density at the lithium nucleus was non-physical. This problem was partially bypassed because their integration radius would have been larger than the radius r c within which the pseudopotential acted (see the ‘Pseudopotentials’ section), and so, they could assume that the integrated charge density was an indication of the hyperfine interaction. In most cases, their calculations display good comparisons to experiment. However, they also acknowledge that their calculations can only be used to predict the sign and rough magnitude of the shifts.
Interestingly, Carlier et al.'s calculations revealed that the previous assignment of experimental Ni peaks in the MAS NMR spectra for 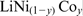 O2 materials, by Marichal et al.,
61
to Ni first and second nearest neighbours of the Li+ should have been reversed. Marichal et al. had used a set of generalised rules to assign the peaks. It had also been assumed that, out of the two competing mechanisms contributing to the hyperfine shift in these materials, the polarisation mechanism was negligible compared to the delocalisation mechanism. The calculations revealed that while delocalisation was important, there was also a significant contribution due to polarisation, leading to hyperfine shifts of opposite sign than previously supposed.
O2 materials, by Marichal et al.,
61
to Ni first and second nearest neighbours of the Li+ should have been reversed. Marichal et al. had used a set of generalised rules to assign the peaks. It had also been assumed that, out of the two competing mechanisms contributing to the hyperfine shift in these materials, the polarisation mechanism was negligible compared to the delocalisation mechanism. The calculations revealed that while delocalisation was important, there was also a significant contribution due to polarisation, leading to hyperfine shifts of opposite sign than previously supposed.
In the last decade, further calculations have been conducted using the CRYSTAL code.78,79 Mali et al. used the PAW formalism to calculate the Fermi contact shift for 6Li in Li2FeSiO4 polymorphs 80 with excellent agreement to experiment (see Fig. 5).
Comparison of calculated Fermi contact shifts (red cross) with experiment by Mali et al. 80 The pseudo-contact interaction is the spin–dipolar interaction expressed in equation (11) (reprinted from Ref. 80 with permission: © 2011 American Chemical Society)
Platinum nanoparticles
Platinum is a particularly interesting system to study using NMR. The only isotope that can be investigated using magnetic resonance is 195Pt, which has a healthy abundance of 33.8 and a moderate gyromagnetic ratio of 5.80446 × 107 rad T− 1 s− 1 (Ref. 81, p. 5), leading to good resonance intensities and therefore sensitivities compared to other nuclei. It has nuclear spin- and therefore no quadrupolar broadening. The dipolar interaction between nuclei and conduction electrons is rapid, resulting in only a very small dipolar broadening (see the ‘Solid state NMR’ section).
82
Most importantly, bulk 195Pt has a very large bulk Knight shift of − 3.4 even at low temperatures.83,82
and therefore no quadrupolar broadening. The dipolar interaction between nuclei and conduction electrons is rapid, resulting in only a very small dipolar broadening (see the ‘Solid state NMR’ section).
82
Most importantly, bulk 195Pt has a very large bulk Knight shift of − 3.4 even at low temperatures.83,82
Measuring how the Knight shift varies as a function of particle size, temperature and surface conditions can be used to investigate the electronic, magnetic and chemical properties of platinum nanoparticles. This knowledge can be used to increase their catalytic efficiency in catalytic converters and fuel cells, 84 and to tailor their properties for electronic devices, gas sensing and medical diagnostics. 85
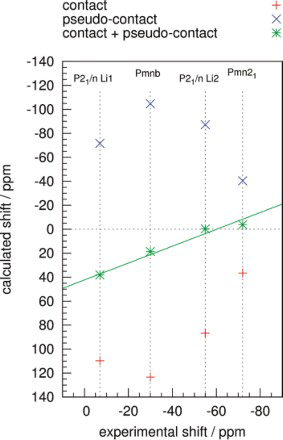
NMR studies on platinum ‘particles’ (diameters 3.3–20.0 nm) date back to the 1980s.86–93 Yu et al. measured the linewidths of 195Pt NMR resonances in platinum particles as a function of diameter, temperature and applied magnetic field.86,87 The Knight shifts were found to be the same as in bulk platinum. The linewidth was independent of temperature but increased with decreasing particle diameter and increasing magnetic field (see Fig. 6). They suggested that the linewidth correlation to 1/diameter could be due to surface effects on the platinum Knight shift since the surface/volume ratio varies in the same way for a sphere. They also found that varying adsorbates on the particle surface (via chemisorption of hydrogen or oxygen) did not affect the spectra.
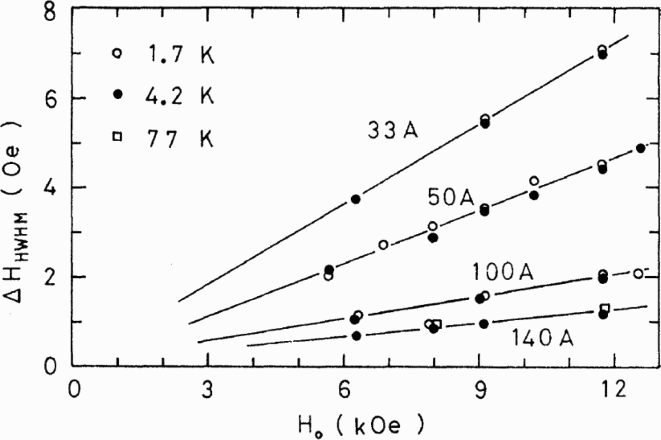
Measurements obtained by Yu et al. 87 showing how the half-width half-maximum of the main 195Pt resonance peak varies as a function of particle diameter (in Å), applied magnetic field (Ho) and temperature (in K) (reprinted from Ref. 87 with permission: © 1980 American Physical Society)
Effect of particle size and temperature on the Knight shift
In general, the NMR spectrum for platinum nanoparticles consists of two peaks. One peak is at 1.138 G kHz− 1, which is the same resonance observed in bulk platinum metal94,95 and is therefore attributed to nuclei in a ‘bulk-like’ environment in the interior of the nanoparticle. The other at 1.100 G kHz− 1 is attributed to nuclei on the surface.84,96
Yano et al. measured the 195Pt resonances on carbon-supported platinum nanoparticles ∼1–5 nm in diameter. 96 Their results in Fig. 7 clearly show these ‘surface’ and ‘bulk’ peaks. The intensity of a peak is proportional to the number of nuclei in that environment. Therefore, the smaller the nanoparticle, the larger the fraction of surface atoms and the higher the intensity of the surface peak at ∼1.100 G kHz− 1. Similarly, the more atoms in the bulk-like interior of the nanoparticle, the larger the intensity of the bulk peak. Even small nanoparticles of 4.8 nm display significant bulk-like character. Conversely, in the smallest nanoparticles of 1.6 nm diameter, there is almost no evidence of any bulk-like behaviour except for some broadening in the surface peak, which suggests that even the few atoms in the interior of the nanoparticle are strongly influenced by those at the surface. The bulk peak present in the nanoparticles of diameter >2.6 nm varies with particle size, with the peak position tending to the bulk metal position of 1.138 G kHz− 1 as the particle size increases. This would appear to contradict the previous conclusion by Yu et al. that the bulk peak position does not vary with diameter.86,87 The majority of Yu et al.'s particles, however, were much larger than those studied by Yano et al., which suggests that the bulk peak tends to the bulk metal value in particles larger than ∼3–5 nm. Furthermore, Yano et al. observed hardly any change in the ‘surface peak’ position as a function of particle size. This implies that surface electronic properties are not affected by particle size (Fig. 7).
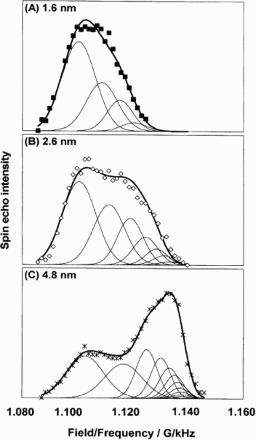
195Pt NMR spectra for nanoparticles. 96 The symbols are the experimental intensities obtained from spin echo experiments on carbon-supported platinum nanoparticles. The thin solid lines are modelled resonances for individual layers and the thick solid lines are the sum of the thin solid lines to give an overall modelled NMR spectrum for each nanoparticle (reproduced from Ref. 96 with permission from the PCCP Owner Societies)
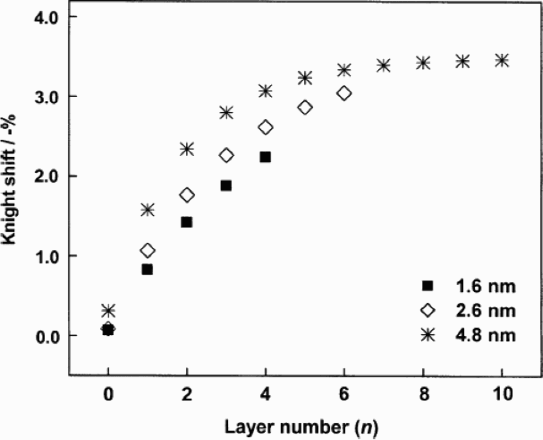
Calculated Knight shifts for each layer inside Pt nanoparticles using layer model analysis. 96 The surface layer is n = 0 (reproduced from Ref. 96 with permission from the PCCP Owner Societies)
Yu et al., to aid the interpretation of their line broadening experiments (see Fig. 6), tried to model the NMR absorption lines.87,86 They assumed that the Knight shift only arose from s-electrons confined within a sphere acting as a free electron gas. The electron and electron spin densities were then calculated. The theoretical model showed good agreement to their experiments (see Fig. 9).
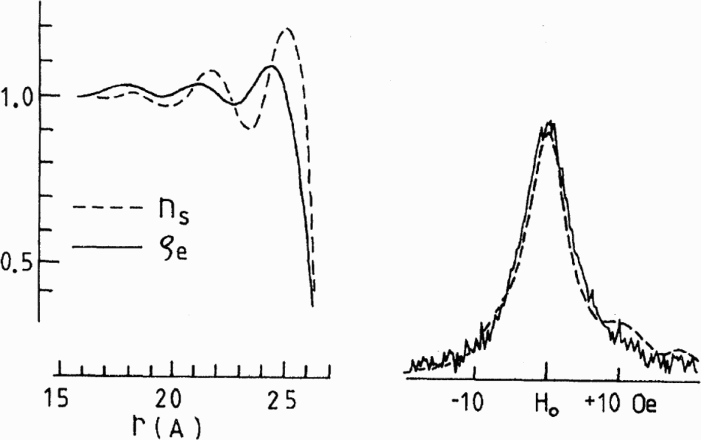
Left: calculated free electron charge density ρe and spin density ns for a particle of radius, r, of 2.7 nm. Right: comparison between the corresponding calculated resonance peak (dashed line) and experimental peak (solid line). The experimental data correspond to a particle of 5 nm diameter measured at 10 kOe and 4.2 K (see Fig. 6) 87 (reprinted from Ref. 87 with permission: © 1980 American Physical Society)
Since the Knight shift for each atom is dependent on its local environment and is related to the electron density at the nucleus (equations (13) and (14)), an approximate NMR peak can be simulated by summing up the local density of states (LDOS) around all magnetic nuclei in the system. In 1993, Efetov and Prigodin employed the supersymmetry method to calculate these LDOS and thus the resonance peaks for nanoparticles independent of composition. 97 They found that the Knight shift decreased, while the broadening increased and became more asymmetric, with decreasing particle size and temperature.
In 1997, Pastawski and Gascón modelled NMR line shapes in particles, independent of composition. 26 They modelled the particles as cubes of atoms arranged in a simple cubic structure. The LDOS for the s-electrons within the layers of these cubes was then calculated using a tight-binding Hamiltonian. Adding the LDOS for the layers together produced a simulated NMR peak. They observed the same trends as Efetov and Prigodin, but additionally found that these trends broke down at very small diameters. In a later paper, Pastawski and Gascón concluded that the breakdown in trend was due to the large surface area/volume ratio of small particles and that the existence of ‘surface states’ would lead to large line broadenings. 98 This is consistent with the ‘surface peak’ apparent in experimental spectra of very small nanoparticles in the above sections. This approach was extended by Fritschij et al. 2 years later. 99
Yano et al. 96 and Tong et al. 84 used a layer model to simulate NMR spectra. They treated the nanoparticle as a set of concentric layers each in a cubo-octahedral arrangement. The NMR signal from each layer was then modelled as a Gaussian distribution. The sum of these Gaussians, weighted by the proportion of atoms in each layer, produced the simulated spectrum for the nanoparticle as a whole. In Fig. 8, the modelled spectra show good agreement to experiment. This shows that the Knight shift in the centre of the nanoparticle varies with particle size; however, the Knight shift of the surface atoms is independent of size. This implies that the surface states are not affected by those in the nanoparticle interior.
The electrons predominantly involved in the Knight shift are situated close to the Fermi level, Ef. Therefore, Yano et al. 96 concluded that resonance peak variation with size occurs as a result of the total change in the s- and d-like LDOS at Ef within each layer.
The hyperfine interaction can be expressed as the sum of positive s-like and negative d-like contributions. 100 In bulk Pt, the d-like hyperfine interaction is the dominant contribution to the Knight shift. However, in a nanoparticle, this d-like component decreases monotonically from the bulk-like centre to the surface.101,102,96 Therefore, the Knight shift decreases from the centre to the surface of the nanoparticle and also decreases with decreasing particle size.103–105
Effect of adsorbates on Knight shift
Tong et al. investigated how different adsorbates (ruthenium, hydrogen, carbon monoxide, cyanide, sulphur and oxygen) alter the 195Pt Knight shift in pure platinum nanoparticles. 84 They found that the experimental surface peak was significantly shifted across a range of 11 000 ppm.
The adsorption of CO decreases the efficiency of platinum catalysts, particularly those in direct methanol fuel cells and hydrogen fuel cells. This CO ‘poisoning effect’ 106 can be combated by alloying platinum (Pt) with ruthenium (Ru).107–109
There are two proposed mechanisms for why Ru reduces the ‘poisoning effect’ of CO in Pt
106
:
Bifunctional mechanism—Ru promotes the creation of ‘oxygen-containing species’, which then react with the Pt–CO surface complex to oxidise the CO from the surface, Ligand field effect—Ru alters the electronic structure of Pt.
The Wieckowski group conclude from measuring the Knight shift in 195Pt-NMR spectra
106
and infrared (IR) data
110
that there are contributions from both mechanisms. The reasoning is as follows (see Fig. 10):
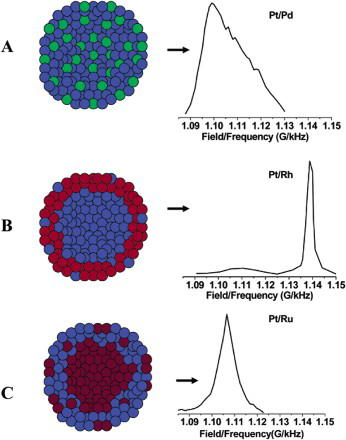
A selection of 195Pt-NMR spectra for different platinum nanoparticle compositions along with representations of their atomic structure 106 . The nanoparticles are 2–3 nm in diameter. Blue = Pt; green = Pd; red = Rh; brown = Ru (Figure and Spectrum C are reprinted from Ref. 106 with permission: © 2003 American Chemical Society; Spectrum A is reprinted from Ref. 111 with permission: © 1996 American Chemical Society; Spectrum B is reprinted from Ref. 112 with permission: © 1988 Royal Society of Chemistry)
The resonance peak in Pt nanoparticles (Pt-black) is subject to a large amount of broadening due to the range in Larmor frequencies from nuclei in the centre of the nanoparticle to those on the surface. The bulk Knight shift decreases with decreasing particle size. For example, the same group measured the bulk peak in 6 nm particles at 1.1322 G kHz− 1, whereas for 2.8 nm nanoparticles, it was at 1.1306 G kHz− 1.
106
Alloyed nanoparticles consisting of a homogeneous solid solution tend to give broader peaks. However, if one of the elements clusters at the surface, then the peak becomes much sharper because there are a reduced number of nuclear environments. An example of homogeneous nanoparticle NMR spectra, in this case for a Pt–Pd nanoparticle, is shown in spectrum A in Fig. 10. Spectrum B in Fig. 10 shows a 195Pt-NMR spectrum corresponding to Pt–Rh nanoparticles. The sharpness of the peak suggests clustering of Pt atoms. This peak is at 1.138 G kHz− 1 (same as the bulk metal), which suggests that this cluster of Pt atoms must be in the centre of the nanoparticle. Therefore, the surface must be enriched with Rh. Spectrum C in Fig. 10 shows the 195Pt resonance peak for Pt–Ru nanoparticles of 50–50 atomic composition. This is a fairly sharp peak, though not as sharp as in the Pt–Rh case. Assuming that the difference in elements does not affect the broadening, this suggests that there is a small degree of segregation (though not as much as in the Pt–Rh nanoparticle). The peak is situated at 1.104 G kHz− 1. Since this is much smaller than the bulk value, this suggests that the surface is enriched with Pt atoms and that the Ru are mostly in the interior. These apparent changes in Knight shift as a function of Pt position suggest that the ligand field affect is a valid mechanism. Additional IR spectra
110
on ‘equivalent’ nanoparticles suggest that the Ru atoms on the surface are not evenly distributed but clustered. IR peaks were present for CO being adsorbed to Pt and also for CO being adsorbed to Ru. Thus, if there are regions of Ru on the surface, there must also exist boundaries between the Pt and Ru regions. These boundaries are necessary for the bifunctional mechanism. Overall, it can be deduced that the structures required for both mechanisms are present in the Pt–Ru nanoparticles. Therefore, it cannot be ruled out that both mechanisms are acting, though it cannot be said that they must necessarily both be acting.
195Pt-NMR, along with a series of heat treatments, was used by Babu et al. on Pt–Ru nanoparticles to conclude that the efficiency of the oxidation of methanol can be raised by heat treating the nanoparticles to ∼200°C. This is because the heat treatment alters the composition of Ru on the nanoparticle surface. 113
In addition to measuring 195Pt resonances, 13C resonances from CO have been measured. These resonances also display a shift that appears to be related to the platinum Knight shift and can therefore be used to obtain information on the bonding environment and mobility of adsorbants on the nanoparticle surface. Babu et al. recorded 13C-NMR spectra for CO adsorbed onto the Pt–Ru nanoparticle surface. 106 This shift was smaller than that for CO adsorbed onto pure platinum nanoparticles (Pt-black). Kobayashi et al. used 13C NMR measurements to further investigate the role of CO on the surface of Pt nanoparticles.114,115 This was continued by Babu et al. 116 with the conclusion that the diffusion of CO on the nanoparticle surface is not the factor that limits the rate at which the nanoparticles catalyse the oxidation of methanol because the surface diffusion of CO is too fast.
There has been some research on how to control the size and shape of platinum nanoparticles since this can be used to tailor their properties. Ramirez et al. systematically investigated the effect of adding the weak ligand HDA (hexadecylamine) to stabilise the surface of the nanoparticle. 85 They recorded the different morphologies formed as a function of varying solvent concentrations of THF and toluene, carbon monoxide and H2, and of changing which synthesis step involved the addition of HDA. They concluded that the strength at which the amine group in HDA bonded to the nanoparticle surface strongly influenced the final platinum morphology. The probability of coalescence is increased by weakening the bonding of the ligand to the surface of the nanoparticle and increasing the mobility of the amine group on the surface. This is because the HDA molecule undergoes a chemical exchange process on the Pt surface with bound (those bonded to the surface) and unbound (those in solution) HDA molecules switching places. This switching means that there are times when the surface is not passivated at all. At these times, the nanoparticles are more likely to bind to one another and thus coalesce. It was also hypothesised that the long chain of HDA molecules acted as a template to guide the nanoparticles into forming nanowires or nanorods.
In the same paper, Ramirez et al. also conducted some solution based 13C-NMR studies in order to investigate the bonding of the HDA (ligand) to the Pt nanoparticle surface. 85 A small shift of no more than 1 ppm was recorded in the peaks attributed to the three carbons closest to the amine group in HDA. Ramirez et al. suggested that this was due to the platinum Knight shift, on the basis that the same shift was not observed for HDA-passivated ruthenium and palladium nanoparticles.117,118 This seems reasonable as it is likely that the HDA molecules are bonded to the nanoparticle surface via the amine group. Thus, these three carbons would be the closest to the platinum nuclei and would therefore experience the largest effect of the spin polarisation in the platinum via a series of hyperfine interactions. Furthermore, platinum Knight shifts are large, as mentioned earlier in this section. Therefore, it is possible that the platinum spin polarisation may affect neighbouring nuclei.
Conclusions
Hyperfine interactions manifest in a variety of materials with important applications, such as lithium intercalation materials for lithium-ion batteries and platinum nanoparticles for catalysis. Solid-state NMR spectra provide an excellent means for measuring these hyperfine interactions and thereby deducing the structure and properties of the materials. However, these experimental spectra are often difficult to interpret without the aid of first-principles calculations.
195Pt-NMR spectra for platinum nanoparticles were simulated through a combination of LDOS and layer methods. For lithium intercalation materials, calculations of electron spin densities allowed the signs and approximate magnitude of hyperfine parameters to be estimated. 48 However, a fully quantatitive analysis could not be conducted due to the difficulty in accurately calculating the charge density at the nucleus. The calculation of hyperfine parameters was improved through the development of the PAW method. 52 This allowed for the reconstruction of physically real valence wavefunctions at the nucleus. When used in conjunction with perturbation theory to calculate the effect of core polarisation on the core electrons, reasonable agreements to experiment were achieved for a set of small molecules and fullerene derivatives.56,57,22 However, the accurate modelling of core polarisation has yet to be fully realised.
Acknowledgements
Thanks goes to J. R. Yates (University of Oxford) and B. J. Morgan (University of Bath) for their useful comments.