
Abstract
This study provides an assessment of the high temperature thermochemical reactions of ilmenite and chromite with sulphur and uses the information to analyse the possibility of selective sulphidation of chrome-bearing spinels as a new route for chromite removal from ilmenite concentrates. The work includes both systematic thermodynamic assessment and targeted experimental investigations. Thermodynamic calculation results studying the effect of reactants composition, temperature and different sulphur sources (H2S and elemental S) showed that chromite can be selectively sulphidised at a controlled atmosphere of partial pressure of oxygen (pO2) below 10−10 atm and partial pressure of sulphur (pS2) above 10−6 atm. The addition of carbon with sulphur was found to be useful for the chromite sulphidation reaction. The optimum quantity of sulphur reactant to carbon was found to be 3: 1 (in mole) for 1 mol of chromite. Sulphidation experiments on a mixture of natural ilmenite and chromite at 1100°C for 5 h using 5%H2S as a sulphur source showed that the ilmenite was preferentially sulphidised first, which was also in a good agreement with the thermodynamic assessment. Sulphidation of a naturally occurring chrome spinel contaminated ilmenite concentrate showed that the degree of weathering also played a role in the sulphidation of the chrome spinel. It was also concluded that H2S is not suitable for selective sulphidation of chrome spinel from the ilmenite concentrate and that tightly controlled pS2 and pO2 conditions are required.
Keywords
Introduction
The production of pigment grade titanium dioxide uses rutile as a feedstock because of its relatively high purity, >95 wt-% TiO2. However, as reserves of the natural mineral decline, pigment producers have turned to synthetic equivalents largely derived from the more abundant TiO2-containing mineral, ilmenite (FeTiO3). Depending on the degree of weathering, naturally occurring ilmenite contains ∼40–65 wt-% of TiO2, however, additional processing is required to selectively remove iron and other impurities in order to produce a synthetic rutile (SR) product of 94–96 wt-% TiO2 purity. The SR is then subjected to a high temperature chlorination process that further removes impurity elements resulting in a high purity TiO2 product (>99·99% TiO2).
The major application of the high purity TiO2 is as a pigment for use in paints or as an ultra-white filler for use in paper and plastic applications. These applications all rely on the unique properties of the TiO2, which include high opacity, whiteness and brightness. Even a small amount of impurities remaining after chlorination, however, will impart colour to the TiO2 and decrease its marketability Therefore, it is vital that the ilmenite feedstock used is largely free of impurities, such as chromium (Cr), magnesium (Mg), and vanadium (V) all of which have the potential to impact on the quality of the TiO2 produced (Fisher-White et al., 2007; Grey, 2002; Pownceby and Fisher-White, 2006). A particular problematic impurity in many ilmenite ores (e.g., east coast of South Africa, Senegal, and east coast of Australia) is chromia (Cr2O3), in which a minor level (<0·05%) in the ilmenite feedstock is sufficient to downgrade the market value of the TiO2 (Grosz, 1987; Harben, 2002; Fisher-White et al., 2007).
Australia has large deposits of ilmenite and associated heavy minerals within a region known as the Murray Basin. The Murray Basin represents the remains of a shallow inland sea, covering an area of 320 000 km2 in Southeastern Australia, straddling the borders of Victoria, New South Wales and South Australia (Pownceby, 2010). The deposits, either coarse-grained strandline or finer grained offshore beach placers, provide a potential source of high titanium bearing minerals, containing roughly 40–60 wt-% ilmenite, 30–40% rutile, and 10–30% zircon (Roy et al., 2000). The rutile and zircon components are separable from the ilmenite via magnetic separation, however; the remaining ilmenite concentrate is invariably contaminated with detrital chromia-containing spinel grains (Pownceby, 2005, 2010). While magnetic separation is usually an effective method for treating ilmenite concentrates containing chromia-rich spinel grains in order to achieve a clean separation and a low-chromia product, this procedure is not effective for the Murray Basin material as there is a considerable overlap in the magnetic susceptibility properties of both mineral phases (Pownceby, 2010).
In recent years, high temperature magnetic roast treatments have been proposed as possible methods for treating chromia-rich spinel contaminated ilmenite concentrates (e.g., Nell and Den Hoed, 1997; Fisher-White et al., 2007). Depending on the initial ilmenite composition, magnetic roasting under either oxidising (Westcott and Parry, 1968; Bergeron and Prest, 1976; Nell, 1999), neutral (Grey and Li, 2001) or reducing conditions (Walpole, 1991; Merritt and Cranswick, 1994; Reaveley and Scanlon, 2001) can be applied. The specific roasting regime employed is primarily determined by the ilmenite chemistry, specifically the Fe2+/Fe3+ ratio, and causes the formation of a ferrimagnetic solid solution between ilmenite (FeTiO3) and haematite (Fe2O3), namely a ferrian ilmenite/titanohaematite (FeTiO3)x(Fe2O3)1−x solid solution. The magnetic susceptibility of the solid solution increases several times higher compared to the original unroasted ilmenite while the magnetic susceptibility of chromia-rich spinel remains unchanged during roasting (Nell and Den Hoed, 1997). The resulting large difference in the magnetic susceptibility of the roasted ilmenite relative to the unreacted chromia-rich spinel allows their magnetic separation. This type of process has been successfully applied to ilmenite concentrates from southern Africa, which also have chromia-rich spinel contamination issues (Westcott and Parry, 1968; Bergeron and Prest, 1976; Nell and Den Hoed, 1997). Similar treatments have been proposed for treating Murray Basin ilmenite concentrates, however, the nature of the concentrates provides some complexities. The ilmenite grains exhibit a broad spectrum of chemical alteration and impurity contents such that no single roasting treatment is applicable to all deposits (Grey et al., 1999, 2003; Fisher-White et al., 2007). Analyses of the spinels also indicate a similar broad compositional and magnetic susceptibility reflecting different source regions of the spinels (Pownceby, 2005). There is therefore currently no commercially operating procedure to separate the ilmenite and chromia-rich spinel phases effectively from the Murray Basin.
Pownceby et al. (2011) recently suggested an alternative approach to separate ilmenite from chromia-rich spinel contaminants. The proposed process involved selective chemical reaction at the surface of chromia-rich spinel grains to induce a change in their surface physical properties which could then be exploited by physical separation processes. They demonstrated that when chrome spinel contaminated concentrates were reacted with sulphur and carbon at 1075°C for 10 h, a preferential reaction between the sulphur and chrome-rich spinel grains occurred. This was characterised by the formation of sulphur-rich rims/coating on the spinel grains as opposed to the ilmenite grains which while metallised, remained largely free of sulphur compounds. The suggestion was that the change in rim composition may make the spinels more amenable to separation through physical methods such as froth flotation, electrostatic separation or by magnetic separation, where the combined sulphidisation/reduction may act to enhance the magnetic properties of the ilmenite (by reducing the Fe3+ to Fe) and at the same time reduce the variation in magnetic susceptibility of the chrome-spinels.
The aim of this study is to further examine the possibility of employing a sulphidising roast treatment for chrome-rich spinel contaminated ilmenite concentrates as a new route for chromia removal from ilmenite concentrates in the Murray Basin. Systematic thermodynamic analyses of the systems Fe–Ti–O–S(–H), Fe–Cr–O–S(–H) and Fe–Ti–Cr–O–S–C(–H) were carried out to evaluate the effect of reactants composition, temperature and partial pressures of sulphur and oxygen. Selected experimental investigations using naturally occurring ilmenite, chromite, and Murray Basin ilmenite concentrates were also performed to confirm the findings from the thermodynamic calculations.
Methodology
Thermodynamic analyses
The thermodynamic analysis was carried out using the thermochemical package FactSage 6·4 with the appropriate optimised compound and solution databases. The FactSage databases used in this study were FactPS (pure compound), FToxide (solid and liquid oxide solutions) and FTmisc (liquid alloys and sulphide solutions). The thermodynamic data of pure compounds in the FactSage system were sourced from standard thermodynamic compilation data of JANAF thermochemical tables and NBS tables of chemical thermodynamics. Solid alloys and solid sulphides were modelled using a sublattice/compound energy formalism solution model. Liquid sulphides and liquid alloys were modelled using the modified quasi-chemical and M*O associate models, respectively (Bale et al., 2009).
The major content of Murray Basin chrome spinel is chromite (FeCr2O4), a solid solution between FeO and Cr2O3, with some aluminium (Al), manganese (Mn) and magnesium (Mg) as substitutional and/or interstitial impurities (Pownceby, 2005). In the thermodynamic analyses, however, pure chromite (FeCr2O4) and pure ilmenite (FeTiO3) were considered for the starting point for the calculations. H2S gas and S (elemental) were considered as sources of sulphur for equilibrium reaction calculations while carbon (C), in a pure graphite form, was also included as many SR processes involve char as a solid fuel reductant. The following thermodynamic assessments were carried out:
Calculation and evaluation of the standard Gibbs free energy (ΔG°) formation of oxides and sulphides of the elements (Fe, Ti, Cr, Mn, Al, Si, Mg) relevant to the ilmenite and chrome spinel ores,
Analysis of the stability of ilmenite and chrome spinel at different partial pressures of oxygen (pO2) and sulphur (pS2),
Equilibrium reaction calculations between chrome spinel and ilmenite with different sulphur sources (H2S, S), with/without the addition of C.
Experimental investigations using H2S
Samples
Three types of samples were used in the current sulphidation study: (1) natural chromite ore, (2) a mixed sample of natural chromite and natural ilmenite (1: 1 weight ratio); and (3) an ilmenite concentrate from a typical Murray Basin strandline deposit contaminated with chrome-rich spinels. The bulk composition of the chromite sample derived from inductively coupled plasma (ICP) analysis is shown in Table 1. X-ray diffraction (XRD) and energy dispersive X-ray spectroscopy (EDX) analysis of the sample indicated that the chromite was homogeneous in the form of a (Fe, Mg)(Cr, Al)2O4 spinel, where Mg and Al were the main substitutional elements for divalent Fe2+ and trivalent Cr3+, respectively. The bulk compositions of the ilmenite sample and the ilmenite concentrate from the Murray Basin deposit are also included in Table 1. The chromia (Cr2O3) content in the natural ilmenite sample was 0·3 wt-% which was significantly less than that for the Murray Basin deposit (4·44 wt-%). X-ray diffraction analysis indicated both samples contained primary (less altered chemical composition) and weathered ilmenite grains; however the proportion of primary ilmenite was higher in the natural ilmenite sample as indicated by the lower TiO2 content. This was confirmed by compositional analysis using energy dispersive spectroscopic (EDS) analyses conducted on individual grains within each of the samples.
Bulk composition of the samples used in the experiments
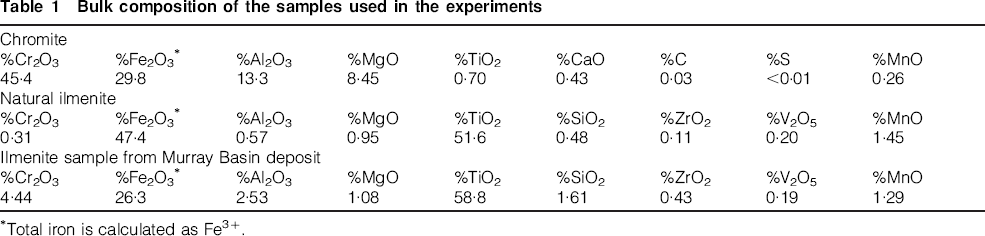
Total iron is calculated as Fe3+.
Sulphidation experiments
Based on the thermodynamic analysis results, experiments were conducted at an isotherm as a means of verifying the calculations. Figure 1 shows a schematic of the experimental apparatus used. For the sulphidation experiments, 1 g of crushed sample, sized between +300 and −425 μm, was placed in an alumina boat (W = 25 mm; L = 100 mm) and located at the hot zone of a horizontal tube furnace. At the beginning of the experiment, the reaction chamber was first evacuated and then filled with high-purity Ar gas through a gas injection rod made of Al2O3. The sample was then heated to 1100°C (±5°C) with a heating rate of 200°C h−1 in an Ar atmosphere. After attaining the required temperature, an Ar–5%H2S gas mixture was introduced into the chamber at a flowrate of 300 mL min−1 and the system was maintained under these conditions for 5 h. Digital mass flow metres were used to maintain the constant flowrate throughout the experiment. After reaching the required time, the chamber was flushed with high purity Ar gas. The sample was then cooled inside the furnace at a cooling rate of 150°C h−1.
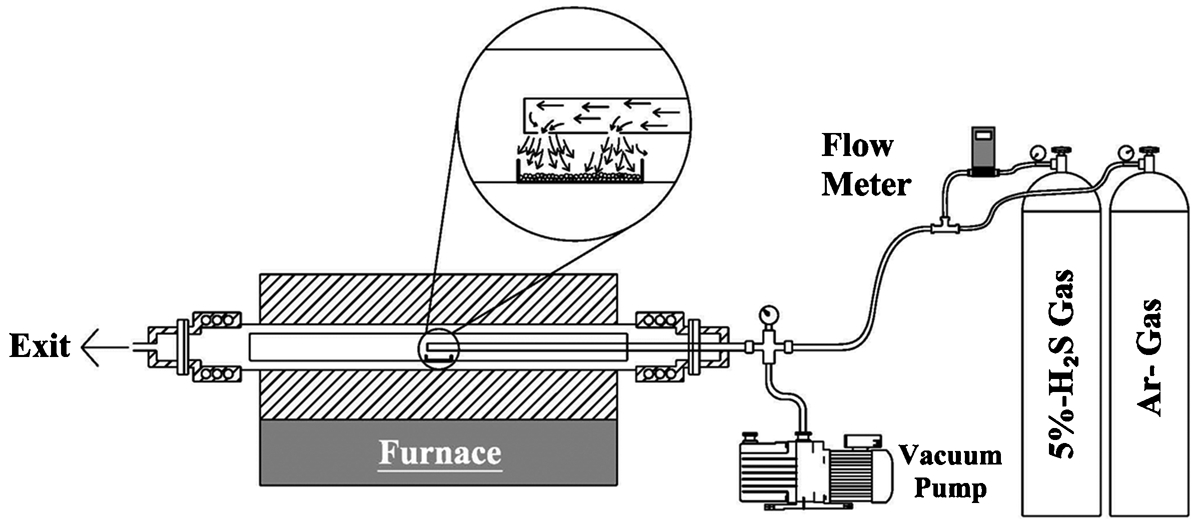
Schematic of the experimental apparatus used for the sulphidation experiments
Analysis
The samples were analysed using various techniques. Scanning electron microscope (SEM) analysis was carried out by using ZEISS SUPRA model 40VP-25-38 Field Emission Scanning Electron Microscope (Swinburne University of Technology) and using a FEI Quanta FEG 400 Environmental Scanning Electron Microscope (ESEM) instrument equipped with a single Bruker XFlash® silicon drift detector for conducting energy dispersive X-ray spectroscopy (EDX). Esprit v.1·9 analytical software (CSIRO) was used to process data obtained. The samples were mounted in epoxy resin and polished up to 1200 grit using SiC paper after which cloth polishing was used with successively finer 6, 5, and 1 μm diamond pastes. The samples were then polished using 0·01 μm alumina suspension until a clean, optically flat surface was produced. The prepared samples were either gold- or carbon-coated by sputtering in vacuum in order to ensure conduction under the electron beam. The accelerating voltage applied for SEM/EDX/BSE imaging ranged between 15 and 20 keV and the working distance was between 6 and 12 mm.
X-ray diffraction was used for phase analysis of the starting and product samples using a Bruker AXS-D8 diffractometer. Approximately 1·5 g of sample was packed into an aluminium sample holder for analysis. Cu Kα radiation (1·54178 Å) operating at 40 kV and 30 mA was used to scan the sample from 20° to 90° (2θ) at rate 0·02° per 1·5 s time step.
Bulk chemical analysis of the ilmenite sample was carried out using X-ray fluorescence (XRF). The sample was finely ground, oven dried at 110°C and then mixed with a flux consisting of a mixture of lithium tetraborate and metaborate. The mixture was then fused in a 95%Pt/Au mould and then cooled using air. The fused disc was analysed on a Philips PW2404 XRF system using an ilmenite-specific algorithm developed by CSIRO.
ICP-AES analysis was used to determine the elemental bulk analysis of the chromite sample. The sample for ICP-AES analysis was prepared using a sodium peroxide fusion procedure and the analysis was conducted at Spectrometer Services Pty. Ltd Coburg, Melbourne, Australia.
Results and Discussion
Evaluation of ΔG° formation of oxides and sulphides
The Gibbs free energy formation (ΔG°) for the different oxides and sulphides were calculated for 1 mol of oxygen (O2) and sulphur (S2) gases between the temperature range 500 and 2000°C. For all calculations, it was assumed that the activity of all elements was 1 at standard state. The ΔG° for FeTiO3 and FeCr2O4 were calculated according to the reactions suggested in the literature (Kale and Jacob, 1992; Ziemniak et al., 2007). The ΔG° variations with temperature for the oxide and sulphide compounds are shown in Fig. 2a and b, respectively.
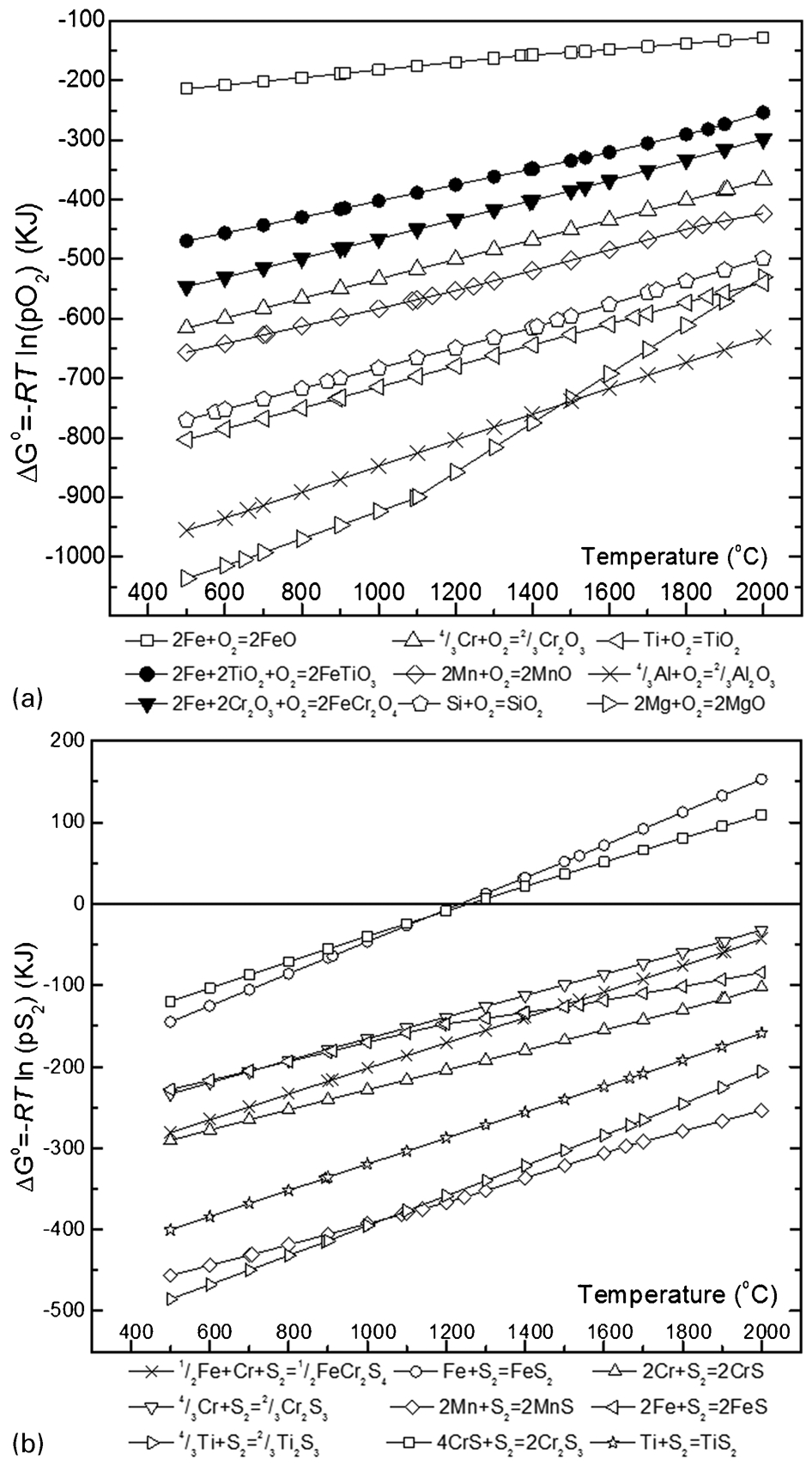
Calculated Gibbs free energy of formation (ΔG°) of various a metal oxides and b metal sulphides
It can be seen from Fig. 2a that the ΔG° of FeCr2O4 is more negative than that of FeTiO3, which indicates that thermodynamically FeCr2O4 is more stable than FeTiO3 in the temperature range studied. Therefore it is expected that during heating in a reducing condition, FeTiO3 will react first from a mixture of FeCr2O4 (chromite) and FeTiO3 (ilmenite). Figure 2a also illustrates the relative stability of different oxide phases (including impurity oxides) in ilmenite concentrates. As FeO is the most unstable oxide compared to others, FeO present within the ilmenite and chromite will react first (being reduced to Fem). Conversely, MgO and Al2O3 are more stable during heating under reducing conditions. For the sulphide system (Fig. 2b), the sequence of most stable sulphide phases is Ti2S3>MnS>TiS>CrS>FeCr2S4>Cr2S3>FeS>FeS2. It has been shown by previous investigators that FeS2 was stable at temperatures below 700°C, however above this temperature, FeS2 dissociates to non-stoichiometric Fe-sulphide and liquid sulphur (Jacob et al., 1979; Kullerud, 1959). From these results, it is anticipated that because ilmenite is less stable compared to both chromite and TiO2, during sulphidising roasting under a reducing atmosphere the ilmenite will dissociate first to FeO and TiO2 and the FeO will then react with the sulphur to form FeS2 (Fig. 2a).
Ilmenite and chromite stability under different oxygen partial pressures
There are only limited studies on the thermodynamic and phase equilibria of the Fe–Ti–O ternary system available in the literature. Merritt and Turnbull (1974) summarised the Fe–Ti–O ternary phase stability data for 1000 and 1200°C and showed that pseudobrookite solid solution of (M3O5) and metallic iron (Fem) formed at high temperatures but became unstable at T<1000°C and transformed into ilmenite solid solution (M2O3) and rutile (MO2) phases. Borowiec and Rosenquist (1981) conducted a study on the system up to 1300°C and determined the pseudobinary sections Fe2O3–Ti2O3 and Fe2TiO5–Ti3O5 while Pesl and Rauf (1999) focused at 1500 and 1600°C. Borowiec and Rosenquist (1981) also carried out experiments using a solid ZrO2 + CaO electrolyte cell for determination of the three phase (iron + wüstite + spinel) combination at different oxygen potentials with temperature. Pesl and Rauf (1999) also detected the presence of a number of phases between TiO2 and Ti3O5 with a general formula of TinO2n−1, where n = 4–20. The study of Itoh et al. (1998) on Fe–Ti––O system at different partial pressures of oxygen with temperature showed the different compositional stabilities of key phases: iron, wüstite, magnetite, ulvöspinel, haematite, ilmenite and rutile.
Seybolt (1960) carried out a phase equilibrium study on the Fe–Cr–O system at 1300°C using Fe–Cr alloy as the starting material under oxidising conditions. They observed the formation of FeCr2O4 spinel at low partial pressures of oxygen (10−13 atm) when the concentration of chromium was higher than 13 wt-%.
Taking all the above into account, in the current study, the stability of ilmenite (FeTiO3) and chromite (FeCr2O4) at 1100°C was evaluated by calculating the equilibrium composition at different oxygen partial pressures from 10−22 to 1 atm. A temperature of 1100°C was selected as this is typical of the operating temperatures used for magnetising roast treatments and for processing ilmenite concentrates to produce SR (Pownceby et al., 2011; Ward, 1990). The calculation was carried out for 1 mol of chromite and 1 mol of ilmenite.
The effect of oxygen partial pressure on ilmenite is shown in Fig. 3a. The calculations predict that at pO2>10−6 atm, ilmenite dissociates to pseudobrookite (Fe2TiO5), rutile (TiO2) and magnetite (Fe3O4). Similar results were derived by Itoh et al. (2006) who carried out oxidising roasting of ilmenite at 1100°C for different partial pressures of oxygen. At lower pO2 (<10−15 atm), metallic iron (Fem) and reduced Ti-oxide phases (e.g., Ti3O5, Ti5O9 and Ti9O17) were formed. All iron from FeTiO3 was converted to metallic iron and no Ti-metal formed. The result was in good agreement with previous results obtained for the reduction of ilmenite with graphite (Wang et al., 2008) and the phase study of the Fe0·394–Ti0·606–S–O system for different pO2 atmospheres previously carried out by Grey and Merritt (1980). Figure 3b shows the Fe distribution in the different phases with changing partial pressure of oxygen. At pO2>10−8 atm it was predicted that the iron phase from ilmenite transforms to Fe3O4 and later to pseudobrookite. At low pO2 (<10−15 atm), iron from ilmenite was predicted to reduce to form metallic iron.
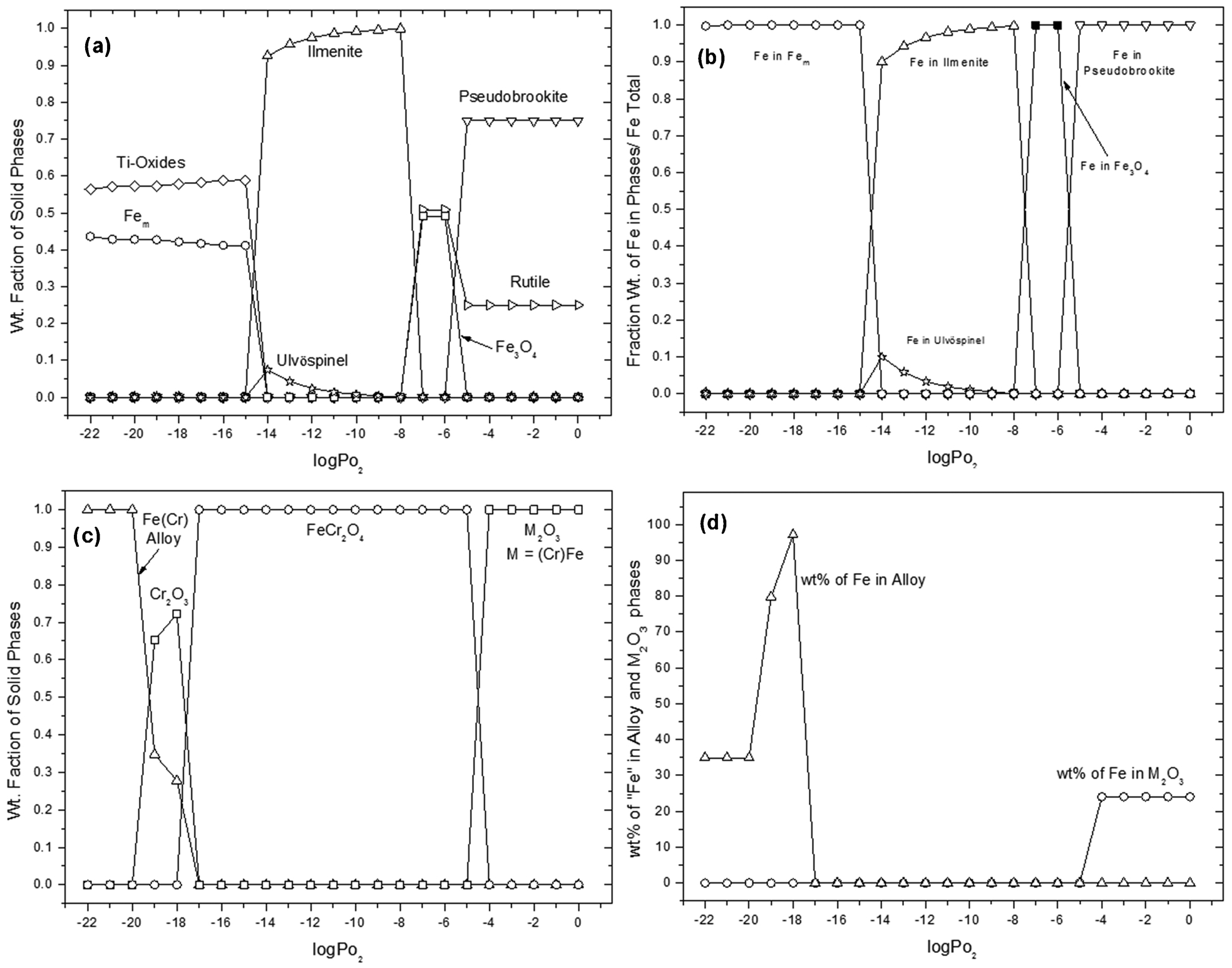
Thermodynamic prediction of ilmenite and chromite stability at 1100°C under different oxygen partial pressures: a plot showing FeTiO3 phase stability, b plot showing the equilibrium Fe distribution among phases in the Fe–Ti–O system, c plot showing FeCr2O4 phase stability, and d plot showing the equilibrium concentration of Fe in the alloy and M2O3 phases in the Fe–Cr–O system
The effect of oxygen partial pressure on chromite (FeCr2O4) stability is shown in Fig. 3c and d. The chromite was predicted to oxidise to the M2O3 (M = Cr, Fe) phase, at pO2>10−4 atm while data in Fig. 3d confirms that the majority of the M2O3 phase is a Cr2O3 compound at high oxygen partial pressure (i.e., only ∼25 wt-% of Fe is present in the M2O3 phase). At low oxygen partial pressures (i.e., below 10−17 atm), some Cr2O3 coexists with an Fe–Cr alloy. Figure 3d indicates that Fe first forms as metal and Cr gradually reports to the metal forming the Fe–Cr alloy.
To evaluate what would happen when a mixture of chromite and ilmenite was processed under various oxygen partial pressures, a comparison of Fig. 3a and c shows that ilmenite is predicted to dissociate to form metallic iron (Fem) and reduced Ti-oxides at pO2<10−14 atm. In comparison, FeCr2O4 is predicted to reduce at pO2 = 10−17 atm which is a significantly lower pO2 compared to the FeTiO3 reduction threshold. This suggests that if a mixture of ilmenite and chromite is reduced at 1100°C under strongly reducing oxygen partial pressures, ilmenite would reduce first, assuming there is no intimate contact between the ilmenite and chromite. The pO2 range for the mixture at 1100°C, whereby the stability of both FeCr2O4 and FeTiO3 is unaffected by the oxygen partial pressure, is therefore predicted to be between 10−14 and 10−8 atm.
Ilmenite and chromite stability at different sulphur partial pressures
There is limited data for Fe–Ti–O–S system relevant to the present study. Grey and Merritt (1980) studied the phase equilibria for the system Fe0·394–Ti0·606–O–S at 1107 and 1212°C at different pS2 and pO2 partial pressures. They found that titanium sulphide would not be expected to be in equilibrium with titanium oxides. They also suggested that, at low pO2, the quaternary system behaved similar to the Fe–S sub-system whereas at low pS2 it behaved like the Fe–Ti–O ternary system. At high pO2 and pS2, the behaviour of quaternary system could therefore be predicted by considering the reactions of the Fe–S and Fe–Ti–O sub-systems and an iron-rich liquid sulphide phase which was formed at higher temperature. More recently, Welham (1998) and Chen et al. (1996) carried out sulphidation experiments on ilmenite with sulphur at room temperature through ball milling action and found pyrite (FeS2) and TiO2 to be the stable phases after milling. Annealing of the products at 600°C showed the decomposition of pyrite to pyrrhotite in both experimental studies.
Similarly, very few data exist in the literature for the Fe–Cr–S–O system. Most of the data available are associated with sulphidation studies of Fe–Cr alloys at selected oxygen partial pressures. Bhogeswara et al. (1979) studied the Fe–Cr–S–O system at 877°C for a fixed oxygen partial pressure of 5·92×10−18 atm and showed the stable phases were pyrrhotite ((Fe, Cr)1−xS), chromite spinel (FeCr2O4), daubreelite (FeCr2S4), Cr2O3 and Cr2S3. Narita et al. (1987) investigated the phase relations and stability fields present in the ternary Fe–Cr–S system at pS2 = 9·87×10−6 and 9·87×10−11 atm at 800 and 900°C and found the stable phases at these conditions were hexagonal crystal structured Fe1−xS and Cr1−xS, spinel (FeCr2S4), and monoclinic Cr3S4. They also found that with decreasing sulphur partial pressure, FeCr2S4 spinel transformed into monoclinic and hexagonal sulphides.
Figure 4 shows the results from FactSage calculations for the phase stability of ilmenite and chromite under different sulphur partial pressures at 1100°C. Only stable solid phases are plotted against sulphur partial pressure. For ilmenite, the pS2 was varied from 3·16×10−2 to 1 atm whereas for chromite, it was varied from 10−10 to 1 atm. The results predicted that chromite was readily sulphidised at a comparatively low pS2 (at about 10−6 atm), while ilmenite sulphidised at the higher pS2 of 1·58×10−1 atm. Ilmenite was predicted to dissociate to matte (FeS) and rutile (TiO2), as shown in Fig. 4a. Figure 4b shows the concentration of Fe in the solid phases. Almost all the Fe reported to the matte and no other condensed phases containing Fe were observed. The chromite was predicted to dissociate to metal oxide (M2O3; M = Cr, Fe), pyrrhotite ((Fe, Cr)1−xS) and a negligible amount of alloy (Fe, Cr) at about pS2 = 10−6 atm, as shown in Fig. 4c. The major metallic phase in M2O3 was Cr, whereas in pyrrhotite Fe was dominant. Although the amount of metallic alloy was very small at low pS2, Fe was the main constituent (Fig. 4d).
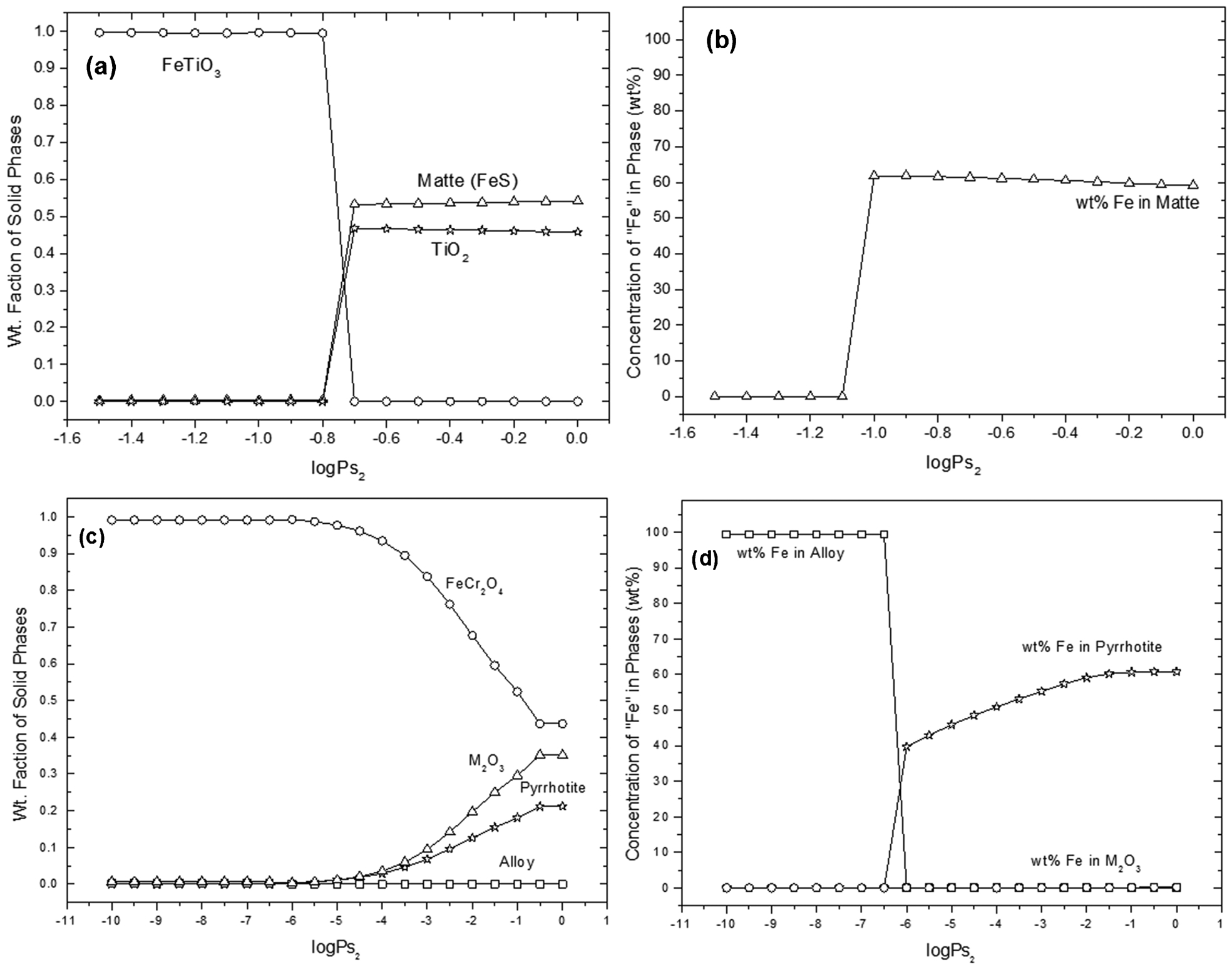
Ilmenite and chromite stabilities under different sulphur partial pressures: a phase compositions, b Fe concentrations in matte for the Fe–Ti–O–S system, c phase composition and d Fe concentrations in stable phases for the Fe–Cr–O–S system
Equilibrium sulphidation reaction of chromite with H2S, S, and C
The sulphidation of chromite using H2S or S and the effects of the presence of C in the system were investigated at temperatures between 450 and 1300°C. Carbon was added into the system as many SR processes involve the use of char as a solid fuel reductant. The calculations were carried out for 1 mol of chromite against different moles of H2S, S and carbon (graphite), a total pressure of 1 atm (set as default), and assuming a closed system. The general reactions investigated are shown below:
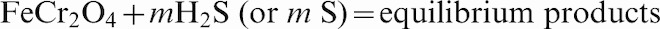
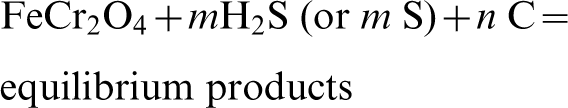
The predicted conversions or amounts of chromite (in percentage) reacted using the different combinations of reactants at different temperatures are shown in Fig. 5. Here, the amount of chromite reacted at equilibrium (η) was calculated from the weight differences of chromite after reaction divided by the initial chromite weight, as per the following equation
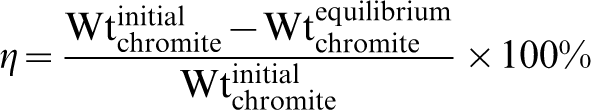
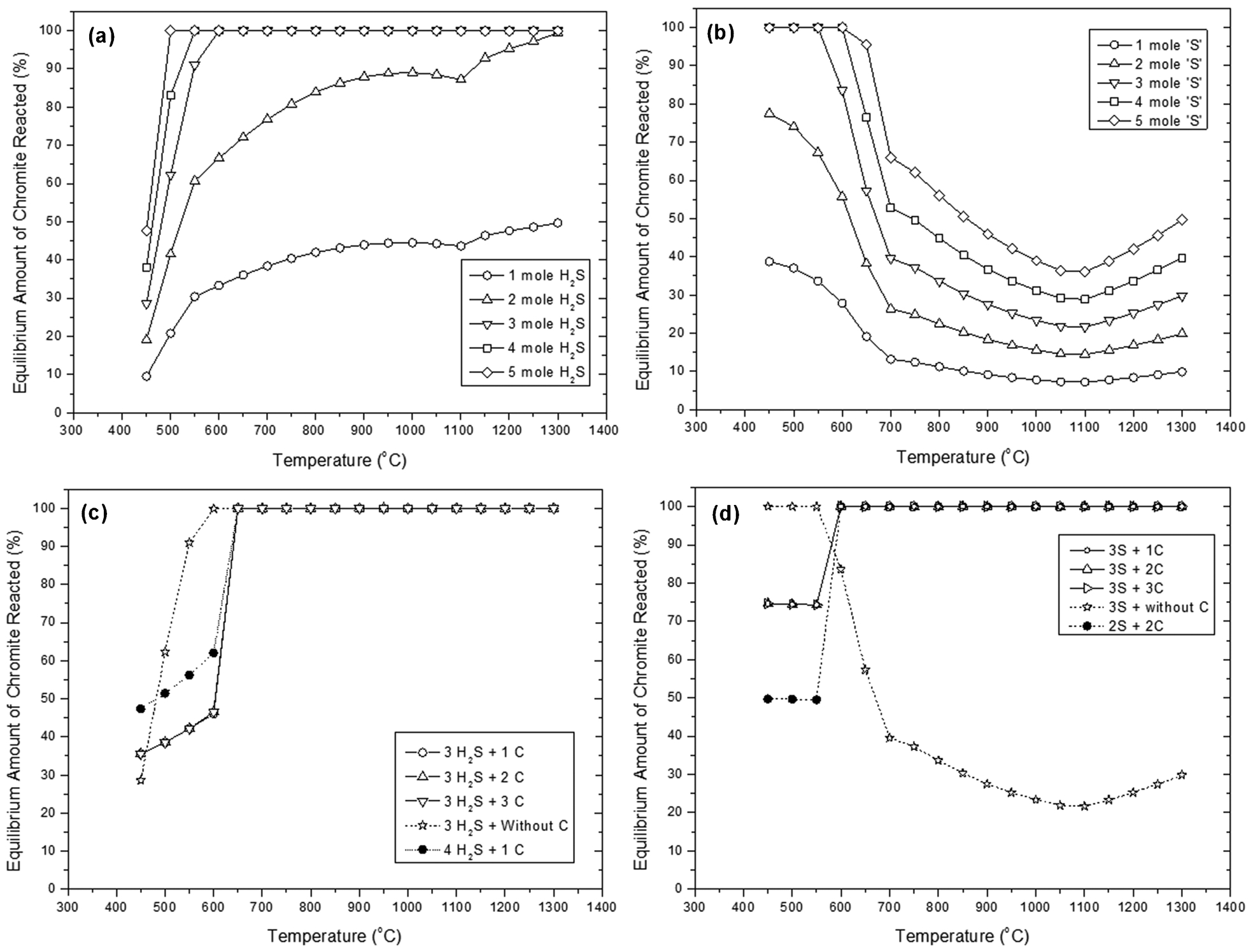
Predicted equilibrium amount of chromite between 450 and 1300°C following reaction between 1 mol of chromite with differing amounts of a H2S gas, b elemental S, c H2S and C, and d S and C
Figure 5a and b shows the equilibrium reaction with different amounts of H2S or S, respectively. In the case of H2S, it can be seen that the higher the concentration of H2S gas, the lower the temperature required for the equilibrium reaction to reach completion. The high concentration H2S meant there was more S available for reaction with chromite. For example, with 5 mol of H2S, maximum conversion could be attained at temperature as low as 450°C while for 3 mol of H2S, maximum conversion was not reached until 600°C. In this scenario, the hydrogen from H2S acts as a reducing agent during reaction, assisting in the breaking of chromite bonds.
In the case of reaction with elemental S, a very different trend was observed. From Fig. 5b, it can be seen that a considerable amount of chromite reverted back at higher temperatures (i.e., T>1000°C). A possible reason for this observation may be that reversion is caused from the oxidising action of SO2 on the FeS2 and FeS which initially forms, resulting in the re-formation of FeO. The Cr2O3 is then able to react with the FeO and transform back to FeCr2O4 as per the following sequence of reactions:
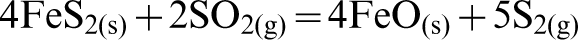
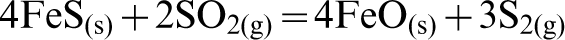
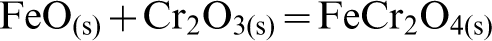
This hypothesis was also supported by the findings of Lee et al. (1983), which described the dual function of sulphur dioxide both as an oxidising and a reducing agent at high temperatures. They also found that SO2 reacted with iron sulphide and formed iron oxide via its oxidising nature.
The effect of carbon addition on the sulphidation with H2S is shown in Fig. 5c. In the presence of C, the maximum conversion of chromite occurred at a higher temperature (650°C) compared to the case with no C (600°C). This is attributed to the reaction of C with S, O and H, forming various gaseous phases and reducing the S available for reaction with the chromite (i.e., effectively reducing the sulphur partial pressure). For example, at lower temperatures (e.g., <650°C), carbon was most likely reacting to produce CO2, CH4 and COS gases as was previously reported by Bhogeswara et al. (1979). Note that the addition of excess amounts of carbon into the system resulted in some carbon left over at lower temperatures.
In comparison, in the case of sulphidation with elemental sulphur, the addition of carbon appeared to have a positive effect on the amount of chromite reacted (Fig. 5d). The presence of carbon appeared to preclude the formation of SO2 hence stopping the reversion back to chromite at high temperatures. It was predicted that the optimum ratio (in mole) of sulphur to carbon was 3: 1 for reaction with 1 mol of chromite at 600°C. Further increase in the carbon amount did not affect the degree of sulphidation at that temperature. Unreacted free carbon was also predicted after adding excess amounts of carbon.
A summary of the equilibrium reaction products predicted from the current study is shown in Table 2.
Predicted equilibrium reaction products from the sulphidation reaction of 1 mol of chromite
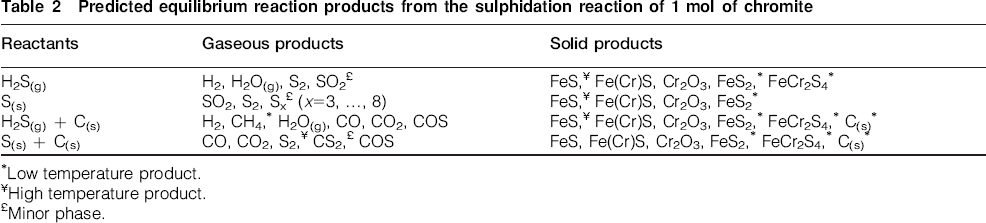
Low temperature product.
High temperature product.
Minor phase.
Equilibrium sulphidation reaction of ilmenite + chromite mixture with H2S, S, and C
Ilmenite concentrates from the Murray Basin are typically contaminated with chrome-containing spinels with varying composition. Typically, the concentration of chrome-spinels in the ilmenite concentrate is between 1 and 5 wt-% (Pownceby, 2005). In order to simulate the sulphidation process of the ilmenite concentrate, equilibrium calculations were performed in which the nominally pure ilmenite and chromite samples were combined (95: 5 in weight respectively) and reacted with different combinations of reactants (i.e., S and H2S, both individually and in the presence of carbon). Carbon was added at a level equivalent to that used in an industrial ilmenite reduction process where it is typically added at a 1: 5 to 1: 10 (charcoal:mineral) wt-% ratio (Cui et al., 2002). The following reactions were investigated:

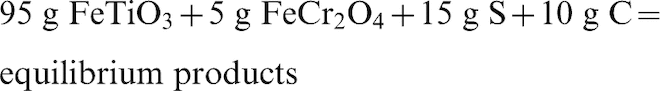
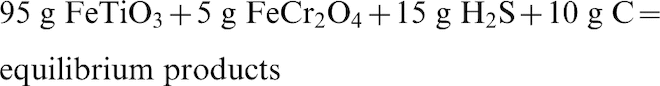
Figure 6a shows the predicted amount of ilmenite and chromite reacted from the sulphidation equilibrium reaction of the ilmenite + chromite mixture with S in the presence/absence of carbon. It can be seen that in the absence of carbon the results are similar to those in Fig. 5b with both the ilmenite and the chromite reacting with sulphur at low temperatures (<600°C). For ilmenite, the results indicated that at high temperatures (above 1100°C) ilmenite dissociates to pseudobrookite whereas at lower temperature (<600°C) it transforms to TiO2 and FeS2 phases. Chromite remained unreacted after an increase in temperature above 600°C. In the presence of carbon, however, about 72% of the ilmenite was readily reacted up to 850°C while no chromite reacted. Above 850°C, more ilmenite was predicted to react until it reached full conversion at 900°C, while the chromite then began to react and reached maximum conversion at 1000°C. The results, therefore, indicate that the addition of carbon with S delayed the complete conversion of both reactants but at the same time assisted in enabling the chromite to dissociate completely. Without C in the system the chromite reacted at lower temperature (<600°C) along with ilmenite, and remained unchanged after a further increase in temperature.
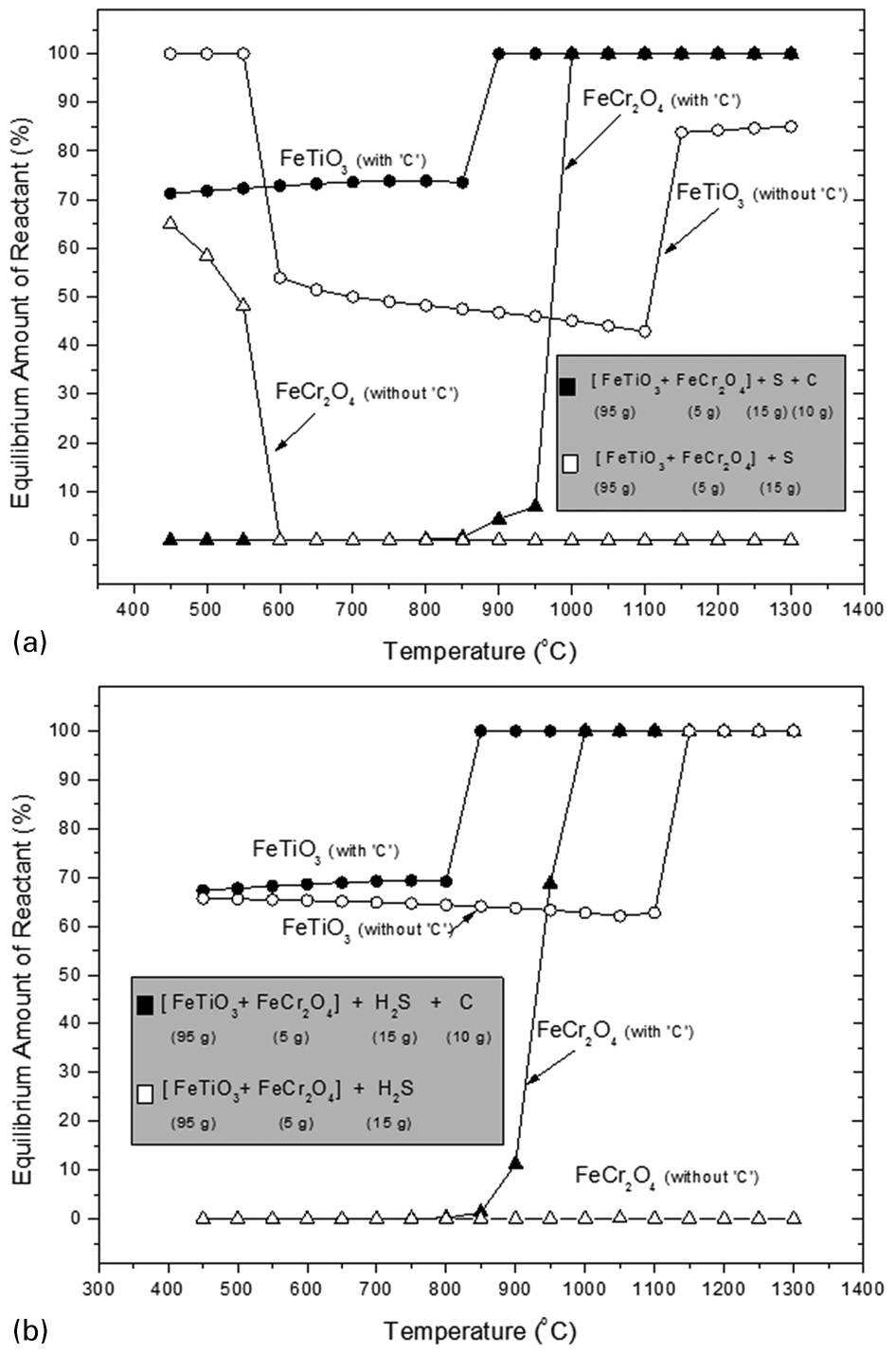
Predicted amount of ilmenite and chromite reacted at equilibrium conditions between a mixture of ilmenite + chromite with a elemental S with/without C and b H2S gas with/without C
The predicted amounts of ilmenite and chromite reacted from the sulphidation reaction using H2S in the presence/absence of carbon are shown in Fig. 6b. In the absence of carbon, it was predicted that about 65% of the ilmenite was readily reacted up to 1100°C. Thereafter, as the temperature was further increased, there was a gradual increase in the amount of ilmenite reacted until it reached maximum conversion at 1150°C. In the absence of carbon, the chromite was not sulphidised at all in the temperature range studied (400–1300°C). When carbon was added to the system, it was predicted that about 69% of ilmenite was reacted at low temperatures (up to 800°C). Under these conditions the chromite also started to react at 800°C, with a gradual increase in the conversion until a maximum was reached at 1000°C. Overall, the results suggest that the presence of C would assist the sulphidation reaction at lower temperatures, both in the case of sulphidation using S and H2S. The results also suggest that ilmenite would react first, before chromite, in both cases, which is expected as the Gibbs free energy of formation of chromite is more negative than that of ilmenite (i.e., the chromite will be more stable than ilmenite, and so not reacting until higher temperatures).
Experimental sulphidation tests
Chromite sulphidation reactions using H2S
Physical examination of the samples produced via the sulphidation reaction between chromite and H2S at 1100°C for 5 h indicated a continuous coating of a brown coloured phase caused by the outer surface of the chromite reacting to form a Fe(Cr)-sulphide compound(s). The XRD analysis results indicated the formation of both Fe(Cr)S and FeCr2S4 type sulphide compounds, as shown in Fig. 7. Scanning electron microscope analysis was also carried out on the sample and the results confirmed the existence of a continuous reaction layer on the surface of the chromite grains (Fig. 8a). Scanning electron microscope examination of a sectioned sample indicated that the sulphide outer layer was continuous and ∼5 μm in thickness (bright rim area in Fig. 8b). The SEM analysis also showed that the outer sulphide layer was underlain by a darker layer/zone ∼15 μm in thickness. Energy dispersive spectroscopic analysis of this layer indicated a depletion in iron compared to the inner core region of the chrome-spinel grain, which remained essentially unreacted. This implies that iron originally present has diffused out and reacted with the H2S to form the sulphide products. It can also be construed from the results that the mechanism of sulphdation of chromite was complex, involving diffusion of selective elements from within the original grains as well as through the dense sulphides product layer. Overall, the results suggest that chromite can be sulphidised using H2S at the conditions studied, in good agreement with the thermodynamic predictions.
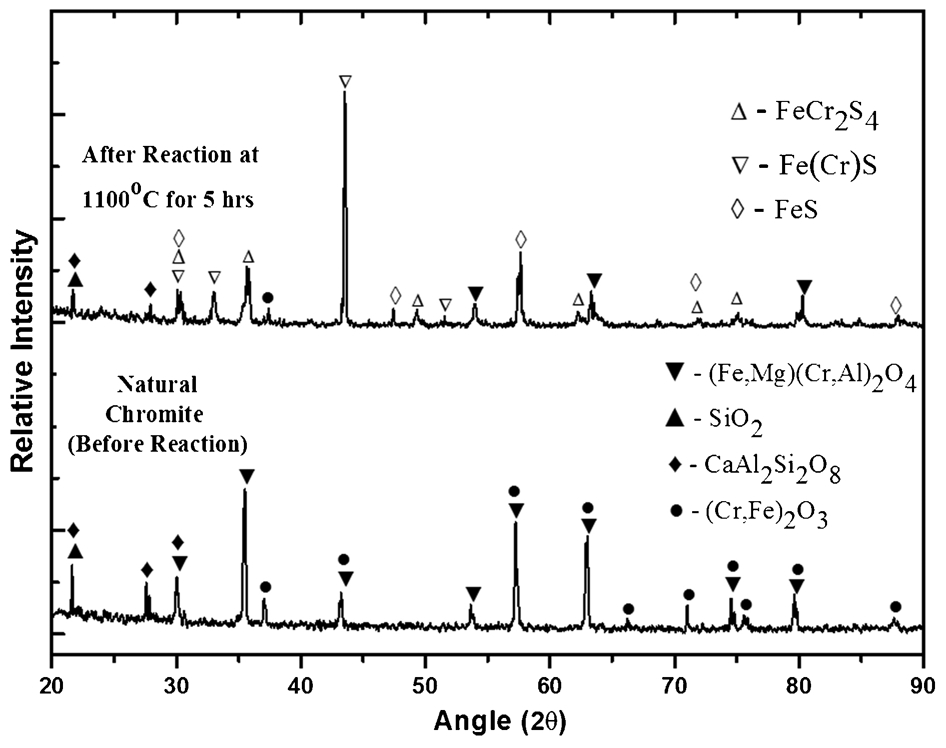
The XRD analysis of chromite sample after sulphidation experiment with H2S at 1100°C for 5 h, showing the formation of Fe(Cr)S and FeCr2S4

The SEM images showing the effects of sulphidation of chromite with H2S at 1100°C for 5 h: a secondary electron image showing the texture of the sample surface and b back-scattered electron image from a sectioned sample mount showing internal textures within the reacted chrome spinel grains
Sulphidation reaction with a mixture of ilmenite and chromite (1: 1) using H2S
A second sulphidation test was carried out using a mixture of chromite and ilmenite grains, physically mixed at a ratio (1: 1) by weight. In this test, equal proportions of both minerals were used for the sake of simplicity of microstructure comparison (i.e., to increase the number of chromite grains available for reaction in the mixture compared to the numbers typically present in a natural ilmenite concentrate). Scanning electron microscope and EDS results from the sample after sulphidation at 1100°C for 5 h are shown in Fig. 9. The results indicate that the majority of ilmenite was sulphidised under these conditions with iron sulphide Fe1−xS observed to form on the surface and within pores and fractures of the ilmenite grains (Fig. 9a). The EDS results in Fig. 9b confirm the formation of the Fe1−xS phase suggesting that the iron from the ilmenite was directly reacting with the sulphur in H2S, forming FeS and TiO2. It is not clear, however, from the current study whether the Fe in the ilmenite was first metallised to metallic iron (Fem), or reacted directly to form the iron sulphide.
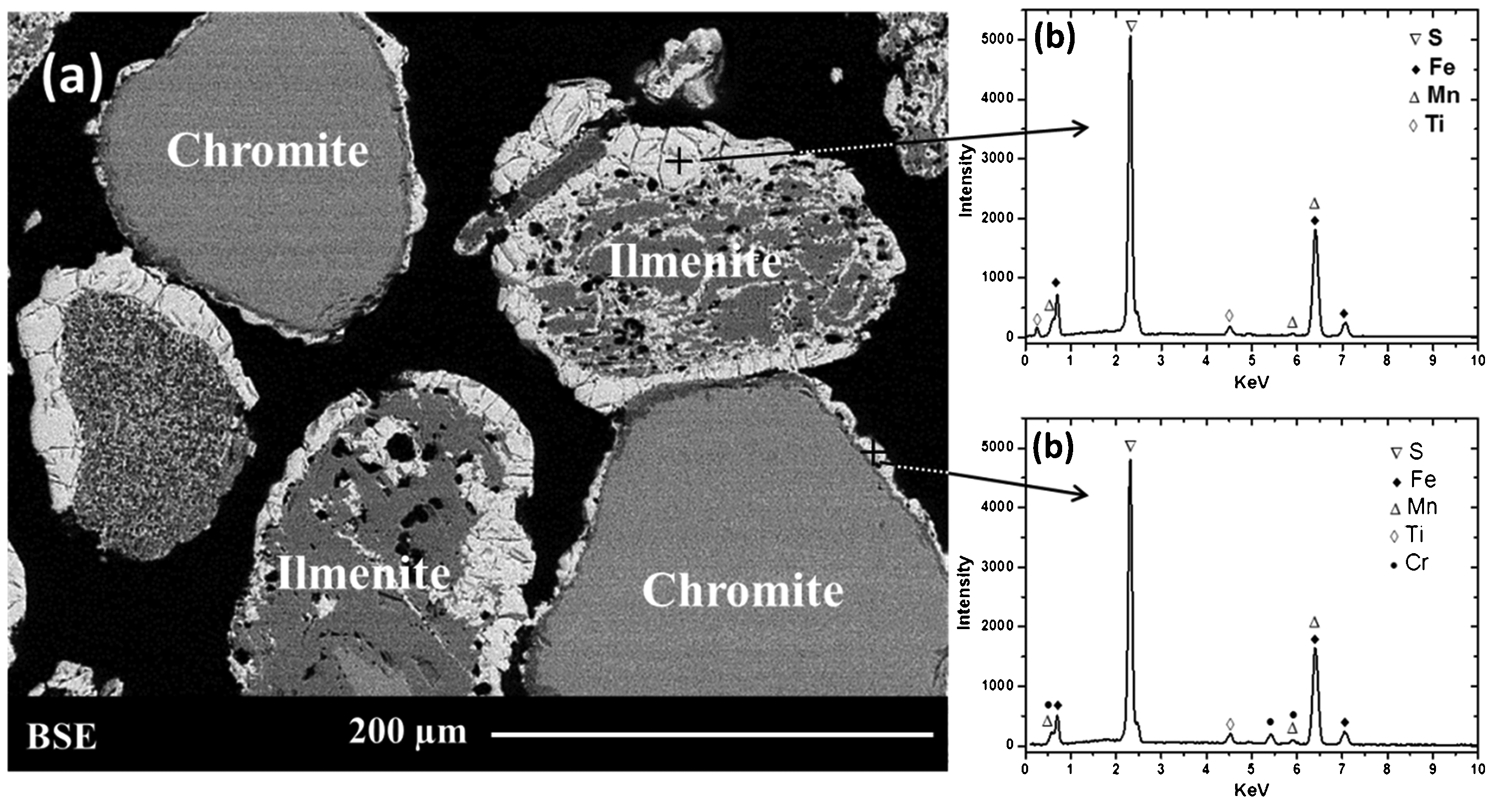
The SEM analysis of ilmenite and chromite mixture (1: 1 weight ratio) after reaction at 1100°C for 5 h: a back-scattered electron SEM image showing the texture of the ilmenite and chromite mixture and (b) EDS showing the elements in the sulphide phases present in the ilmenite and chromite
Sulphidation of the chromite was also observed in the mixture sample as was previously noted in the natural chromite-only experiments. The reaction appeared to be limited to within the outer 15 μm of individual chromite grains and no reaction products were observed within the grains (c.f. the ilmenite results where reaction occurred throughout the grains). It appeared that the denser nature of the chromite limited the reaction on the surface only, and that further reaction would require diffusion of reactants through the diffusion layer and the outer sulphide products. Energy dispersive spectroscopic analysis of the sulphide phase on the surface of chromite grain (Fig. 9b) indicates a trace of Cr present in addition to mainly Fe and S. The latter suggests the likely formation of a pyrrhotite ((Fe, Cr)1−xS) phase at the surface. It can also be observed from Fig. 9a that some chromite grains were bonded or fused with ilmenite grains through the sulphide phases present on the surface.
The XRD analysis results after reaction are shown in Fig. 10 and confirm the formation of the sulphide phases where characteristic diffraction peaks for Fe(Cr)S and FeCr2S4 were detected. Overall, the observed results are in good agreement with the FactSage predictions in the previous sections that ilmenite is more reactive than chromite under these experimental conditions.
The XRD analysis of ilmenite and chromite mixture after sulphidation experiment with H2S at 1100°C for 5 h, showing the formation of sulphide phases
Sulphidation reaction of Murray Basin ilmenite concentrate using H2S
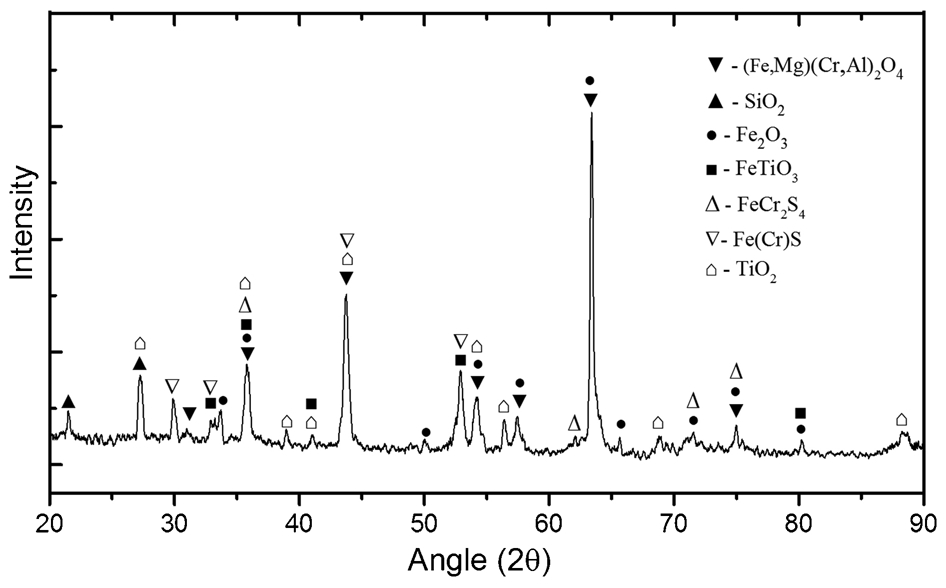
The Murray Basin ilmenite concentrate was sulphidated at 1100°C for 5 h using 5%H2S. Scanning electron microscope/energy dispersive spectroscopy elemental mapping analysis results are shown in Fig. 11. These show that both ilmenite and chromite were sulphidised however there was an excess amount of sulphides around the chromite grains (red grains in Fig. 11). In this case, sulphidation also occurred at the centre of chromite grain through pores and fractures as well as on the outer surface indicating that the weathering degree of the chromite grains played a significant role in the nature and degree of the sulphidation. The average sulphide rim thickness on the surface was also higher than previously observed in the sulphidation of natural (denser) chromite (e.g., Fig. 8b). Energy dispersive spectroscopic analysis of the outer-rim composition confirmed the presence of sulphur and iron, suggesting the formation of iron sulphide phases. Similar to the observations in the experiments with natural chromite, it appeared that Cr ion remained relatively immobile in the grain (as the oxide) while Fe rapidly diffused out to react with sulphur. For the ilmenite grains, sulphidation occurred both on the surface and in the interior of the grains. The XRD analysis is shown in Fig. 12 and confirms the presence of other compounds including TiO2 (secondary rutile formed through reduction of the ilmenite), FeS, unreacted Cr-spinel and magnetite.
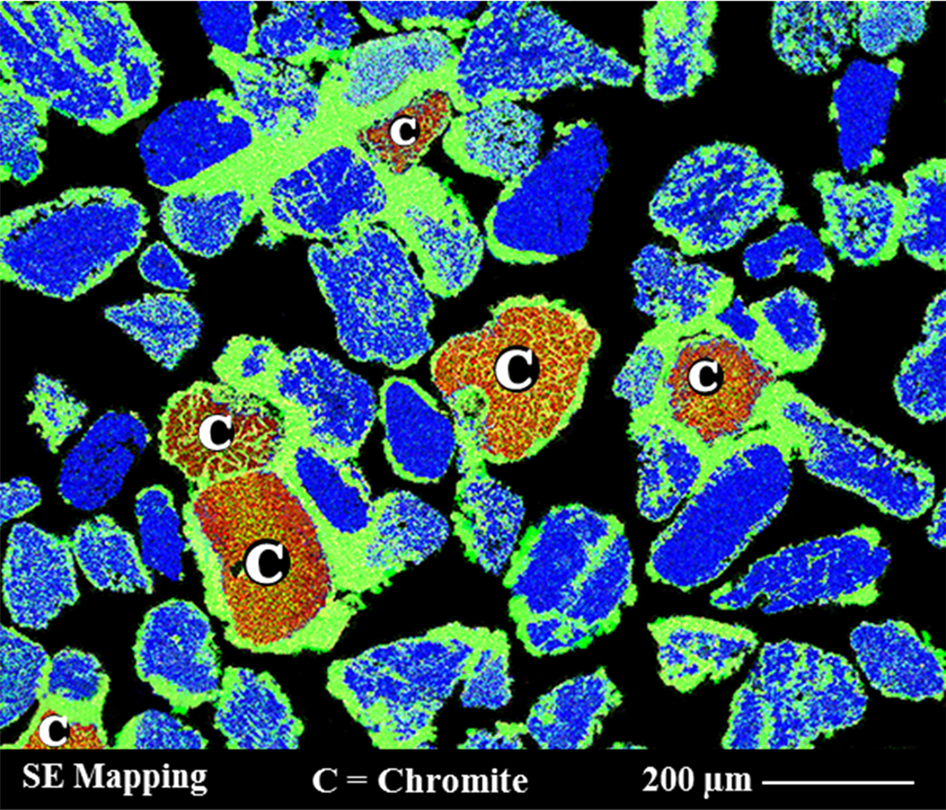
The SEM/EDS elemental mapping of Murray Basin ilmenite after reaction at 1100°C for 5 h showing the chromite reacted grains surrounded by sulphide compounds
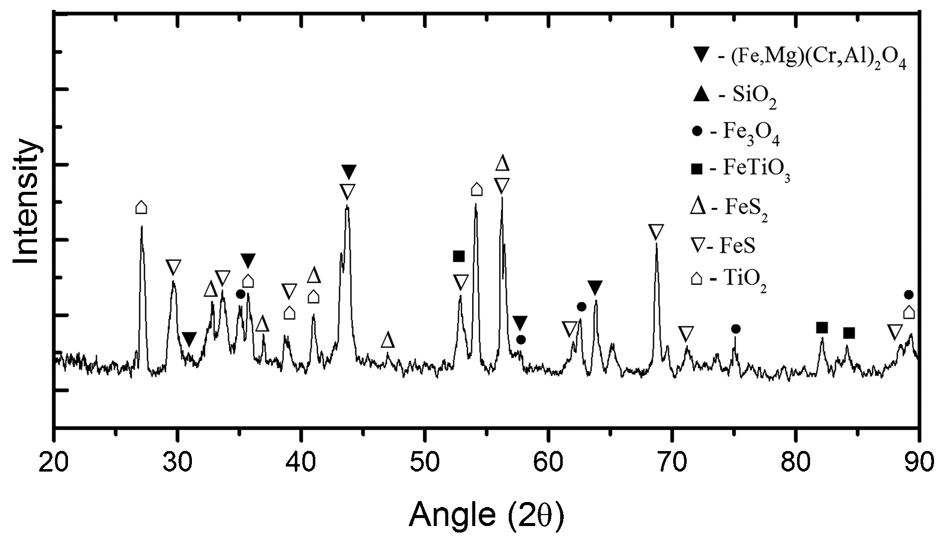
The XRD analysis of Murray Basin as received samples after sulphidation at 1100°C for 5 h showing formation of different sulphide compounds
Overall, it can be seen from the experimental investigations that at the conditions studied, i.e., 1100°C, 5 h, using H2S as sulphidising agent, both the chromite and the ilmenite were readily sulphidised. The chromite could not be selectively sulphidised from the Murray Basin ilmenite concentrate. Even when a diluted H2S (5%H2S–Ar) gas mixture was used, it appears that the gas conditions provide condition which are too reducing and sulphidising. Therefore, the use of H2S gas as sulphidising agent appeared to be not suitable for the selective sulphidation of chromite.
Interestingly, the above analyses (both thermodynamic assessment and experimental) are not in agreement with the results of Pownceby et al. (2011) which showed that under Becher conditions (i.e., 1075°C, presence of S and C) there was selective sulphidation of chromite. This therefore suggests that there must be some operating regime with a specific pO2 and pS2 condition that allows for the selective sulphidation of chrome spinel and not ilmenite (which becomes metallised, but does not contain significant sulphur). Therefore as a part of further analysis, an overlay of predominance phase stability diagrams of the Fe–Cr–O–S and Fe–Ti–O–S systems at 1100°C with changing pO2 and pS2 was also developed and is shown in Fig. 13. From this plot it can be seen that there is a narrow window of operation in which selective sulphidation of chromite grains can potentially be carried out, shown as grey area in Fig. 13. In this window, FeTiO3 and Fe2TiO4 (from ilmenite – dotted line); and FeCr2O4 and FeS (from chromite – solid line) are the stable phases. Thus, when a chromite sample (for example a solid solution of magnetite and Fe2Cr2O4) is subjected to these pS2 and pO2 conditions, one could expect that the iron in the chromite solution may be sulphidised first rather than the iron present in the titanate.
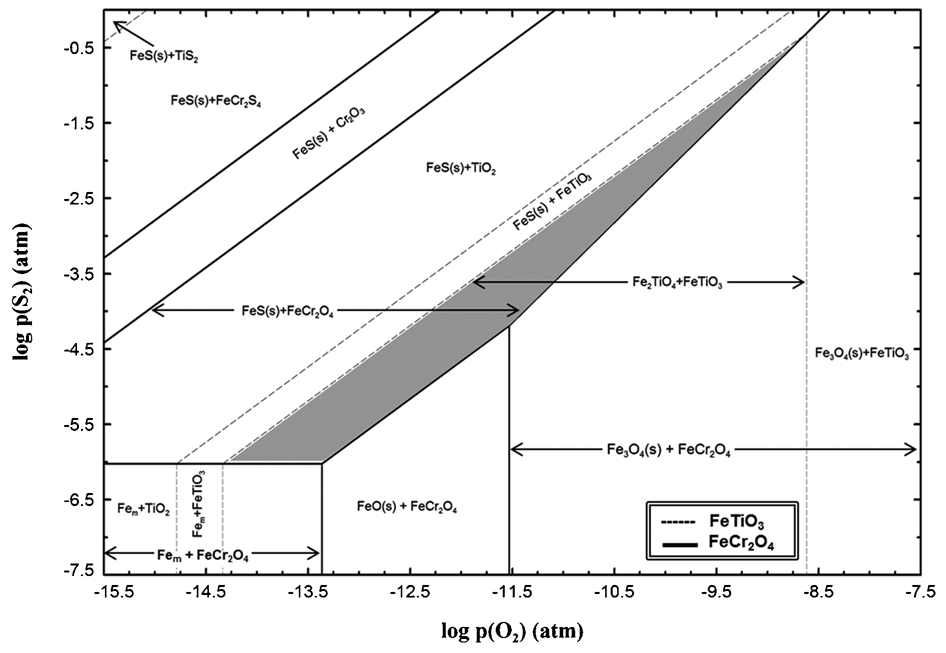
Predominance diagram of Fe–Cr–Ti–S–O system at 1100°C
To clarify further whether there is a window of operation where chromite can be selectively sulphidised compare to ilmenite; additional equilibrium calculations were carried at various pO2 and pS2 using the following general equation
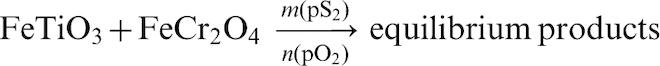
In these calculations, potential solid solution between phases was considered. It was confirmed from the equilibrium calculations that there is a window of operation at pS2 = 10−4·5 and pO2 = 10−13 atm where selective sulphidation of the chromite occurs. Under these conditions, the resultant equilibrium products would be as follows
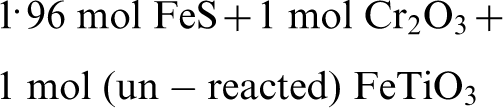
Further investigation at controlled pS2 and pO2, for example using a mixture of CO, CO2 and SO2, will need to be carried out to elucidate the observation of Pownceby et al. (2011) in the context of the results in this study. It should also be noted that the analysis presented above was heavily based on the results from the thermodynamics assessment, which provide the limiting conditions and not considering the kinetics factor. In actual, particularly when low grade (weathered) ores are used, the kinetics factor can play a significant role. Both ilmenite and chromite are thermodynamically susceptible to sulphidation under the studied conditions and the reactant supply is adequate, in this situation the kinetic competition may have a large contributing factor to the ilmenite sulphidation.
Summary
Combined thermodynamic and selected experimental studies on the assessment of selective reduction and sulphidation of chromite grains contaminating ilmenite concentrates have been carried out. From the thermodynamic assessment, the following can be concluded:
The Gibbs free energy of formation of chromite is more negative than that of ilmenite which indicates the higher stability of chromite. At low pO2<10−14 atm, ilmenite was predicted to reduce to various Ti-oxides and metallic iron whereas the chromite was not reduced until pO2<10−17 atm. Ilmenite started to sulphidise at pS2 = 1·58×10−1 atm, while chromite was unaffected until a pS2 = 10−6 atm was reached.
The results predicted that at high temperature (T>700°C) the sulphidation of chromite using sulphur resulted in a lower yield compared to sulphidation using H2S. The addition of carbon appeared to slightly lower the minimum temperature at which the maximum yield was achieved for both reactants. The effect of carbon addition however was more pronounced in the case of sulphidation using sulphur. A sulphur to carbon ratio of 3: 1 (in mole) was found to be optimal for sulphidation of 1 mol of chromite at T = 650°C.
Equilibrium calculation results for the sulphidation of an ilmenite and chromite mixture indicated that ilmenite was more unstable at the conditions studied (T>850°C) and reacted at lower temperatures than chromite (>1000°C). Addition of carbon with H2S appeared to be beneficial helping to dissociate the chromite.
Experimental sulphidation tests of natural chromite using 5% H2S at 1100°C for 5 h showed that sulphide products formed on the surface of the chromite grains. Solid sulphide rims with an adjacent underlying Fe-depleted diffusion layer and unreacted chromite core were observed.
Experimental sulphidation tests using a natural ilmenite + chrome-spinel contaminated Murray Basin concentrate showed that the ilmenite was significantly more reactive that the chrome-spinels being converted to iron sulphide and reduced rutile phases.
In the case of sulphidation of the Murray Basin concentrate, the sulphide products were formed on the surface and interior (pores and fractures) of both the ilmenite and the chromite grains. This was not observed in the sulphidation of natural chromite, where the sulphide product was observed only on the surface of grains. It is likely that the degree of weathering (i.e., enhance porosity and fracturing of grains) plays a vital role during sulphidation of the Murray Basin concentrate allowing the sulphidation of the grains interior.
It appeared that H2S provides both strong reducing and sulphidising conditions such that selective sulphidation of chromite was not achieved. A more strict control of pS2 and pO2 is required.
Footnotes
Acknowledgement
This study is funded by the Swinburne University Postgraduate Research Award (SUPRA) and CSIRO Flagship Collaboration Fund scholarship through the Minerals Down Under National Research Flagship.