
Abstract
New insights are presented on the speciation of surface oxide phases on fine inert gas atomised (GA, <45 and <4 μm) and water atomised (WA, <45 μm) stainless steel AISI 316L powders. X-ray photoelectron and Auger electron spectroscopy, scanning electron microscopy, Raman spectroscopy, and cyclic voltammetry were applied for the characterisation. Oxidised manganese was strongly enriched in the outermost surface oxide of the GA powders (13 and 47 wt-%), an effect increasing with reduced particle size. Manganese and sulphur were enriched in oxide nanoparticles on the surface. Oxidised silicon (59 wt-%) was enriched on the WA powder surface. Tri- or tetravalent manganese oxides were observed on the GA particles in addition to α-Fe2O3, and Cr2O3. The oxide of the WA powder revealed in addition the likely presence of a silicate rich phase, mainly consisting of tetravalent Si, di- and/or trivalent Fe, and hexavalent Cr, which was confirmed not present as chromate.
Introduction
Characterisation of the surface oxide of atomised powders may assist to improve densification during further processing (e.g. sintering, high isostatic pressing, micrometal injection moulding). Especially alloying elements of high oxygen affinity, such as Cr, Si and Mn, may cause some difficulties during further sintering steps.1
Previous findings show that the surface oxide (composition, phases/inclusions and their distribution, thickness, etc.) of metal powders is dependent on the cooling rate during atomisation,2 the particle size and secondary dendrite arm spacing (both connected to the cooling rate),2– 5 the type of atomisation (e.g. water, gas atomisation, or rotating electrode process),4 and the oxygen availability.6 It is generally reported that minor elements with higher oxygen affinity, such as Mn, Si and Cr, are enriched at the surface, an effect that increases with decreasing particle size.1,3,4,6,7 These minor elements may be present as oxide particles or islands within, or on, a more continuous oxide layer. Literature findings report Cr2O3 with MnO and Fe2O3 to be the main components of this layer on nitrogen atomised austenitic AISI 304L stainless steel powder,7 possibly also with MnCr2O4, FeCr2O4 or (Fe,Cr)2O3 present as particles or minor locally occurring surface species.8
Electrochemical identification of metal oxide species in, or on, particles is possible by using cyclic voltammetry. The carbon paste electrode (CPE) has been widely used to identify different metal oxide/hydroxide phases in/on metal-containing powders.9– 14 An advantage of electrochemical analysis compared with surface analytical measures is the direct information on reduction/oxidation processes, important information for considerations of the oxidation state of metal phases of interest. Information on the oxidation state in the surface oxide is essential for toxicological considerations related to occupational hazards, as the oxidation state is one key factor for toxicity for many metals.15 It is also important for electrochemical, catalytic, and chemical (e.g. dissolution) properties of the powders.16
This paper focuses on the multianalytical (XPS, AES, Raman spectroscopy, and cyclic voltammetry) characterisation of surface oxides of gas atomised and water atomised AISI 316L stainless steel powders. The same gas atomised powders have previously been investigated in several in vitro 16– 21 and in vivo 22 tests, combining toxicology, metal release investigations, and particle characterisation.
Materials and methods
Materials
Bulk alloy compositional information for AISI 316L inert gas atomised 316L powder sized <45 μm (GA<45 μm, produced by ANVAL, Torshälla, Sweden), inert gas atomised 316L powder sized <4 μm (GA<4 μm, produced by Sandvik Osprey Limited, UK), and water atomised 316L powder sized <45 μm (WA<45 μm, produced by Höganäs AB, Sweden) is given in Table 1. All powders were supplied via the European Confederation of Iron and Steel Industries and International Stainless Steel Forum, Belgium. The specific surface areas, investigated by the BET method, as described previously,17 are 0·700 (GA<4), 0·069 (GA<45), and 0·087 (WA<45) m2 g−1 respectively.
Bulk and relative surface mass composition (wt-%) of gas and water atomised 316L powders, and observed XPS binding energies*
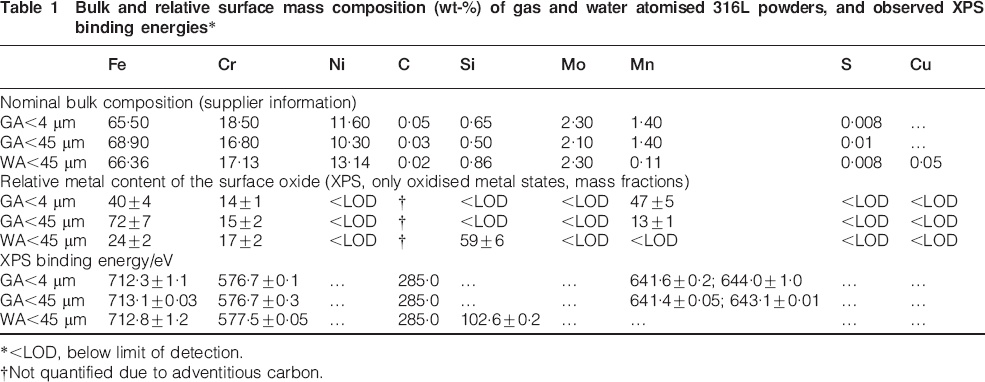
*<LOD, below limit of detection.
†Not quantified due to adventitious carbon.
Field emission gun scanning electron microscopy (FEGSEM), as previously described,23 was used for morphology studies. Quantitative surface oxide investigations were conducted by means of X-ray photoelectron spectroscopy (XPS) and Confocal Raman microscopy (CRM) to identify surface oxide phases. Experimental details are given elsewhere.16 Auger electron spectroscopy (AES) (ThermoScientific MicroLab 350) was used to analyse oxides on GA <45 μm with ∼20 nm lateral resolution. Sensitivity factors from the manufacturer were used for apparent quantification. Ar+ etching at nominal rates as calibrated on Ta2O5 was done to study the distribution of elements in oxide nanoparticles. Owing to their irregular shape, further affected by the etching, the estimated effect of the surface orientation on the etch rate was approximate.24 Cyclic voltammetry [starting at open circuit potential (OCP), polarising cathodically to approximately −1·2 V and consecutively polarising anodically to approximately +0·4 V] was employed to obtain reduction/oxidation information. All measurements were performed with a scan rate of 0·5 mV s−1 in 8M NaOH using a carbon (graphite) paste electrode (CPE) and a reference electrode of Hg/HgO. Experimental details are given elsewhere.25 The concentrations of powder in the graphite paste was 11·6 mg (WA<45 μm), 6 mg (GA<4 μm), and 20 mg (GA<45 μm), added to 100 mg graphite powder. Different concentrations were tested and the difference in BET area was accounted for in order to obtain representative spectra. The concentration of the powder has previously been found to significantly influence the spectrum.25
Results and discussion
Figure 1 shows the morphology of the inert gas and water atomised 316L powders. Previous findings propose the initial formation of oxide particles with strong oxide formers such as Mn, Cr and Si,1,6,7,26 and the subsequent formation of an iron oxide6,7,26 and/or chromium oxide7 layer (and in the case of water atomised particles an oxidised silicon layer27) during the atomisation process and handling. Oxide particles rich in Mn, Fe and Si, were reported by means of SEM for water atomised particles.1 Particles were observed at the surface on the powders sized <45 μm of this study (Fig. 1d). These surface particles, sized ∼20 nm up to a maximum size of 100 nm, were relatively homogeneously distributed on the GA powder particles. For the WA powder, less abundant surface particles were observed, cf. Fig. 1g, compared to the GA powder, suggesting that most of the oxidised silicon (∼59 wt-%, Table 1) was present within the surface oxide layer. No surface oxide nanoparticles were observed on the GA<4 μm powder (data not shown).
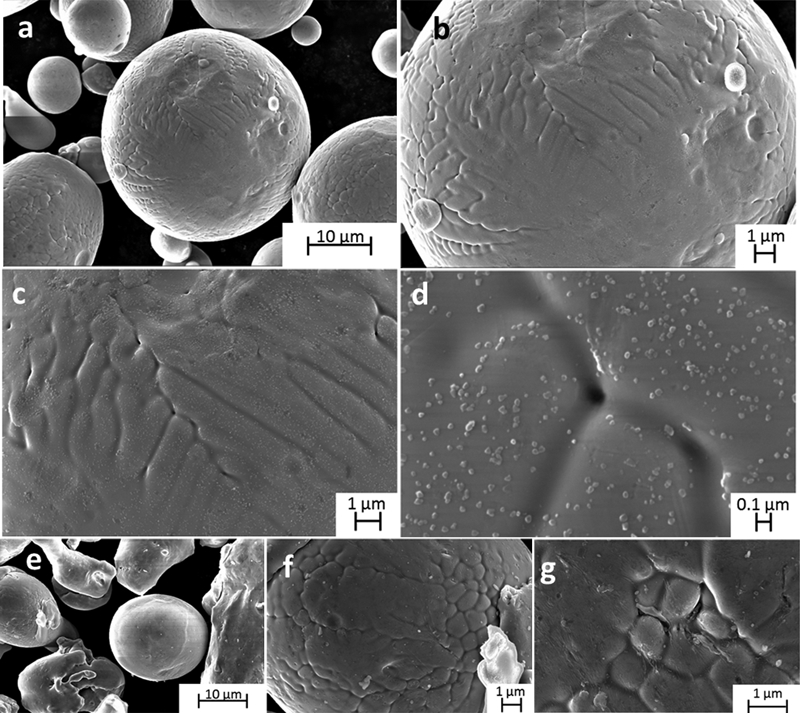
Images (FEGSEM), at different magnifications, of gas atomised (a–d GA<45 μm) and water atomised (e–g WA<45 μm) 316L powders
To investigate the composition and distribution of elements within these nanoparticles compared to the matrix surface, AES point analysis were conducted before and after etching of the GA<45 μm particles (Fig. 2). Both Mn and S were enriched in the oxide nanoparticles compared with the matrix surface (only Fe and O on the outermost surface) (Fig. 2c and d). The relative amount of Mn was similar or less than the amount of Fe in the oxide particles. This was reproducible for all oxide nanoparticles (apart from a few Si rich particles of different morphology and larger size). When assessing an approximate depth profile down to 40 nm by etching and stepwise analysis, it was evident that both Mn and S, as well as Fe existed throughout the entire profile, although in amounts decreasing with increasing depth (Fig. 2d–f). Cr and Ni were detected first at some nm depth. The matrix oxide thickness was estimated to ∼2·5 nm based on depth profiling data (data not shown). The thickness of the oxide nanoparticles was significantly thicker compared to the matrix oxide, however difficult to estimate exactly due to their differences in size and shape.
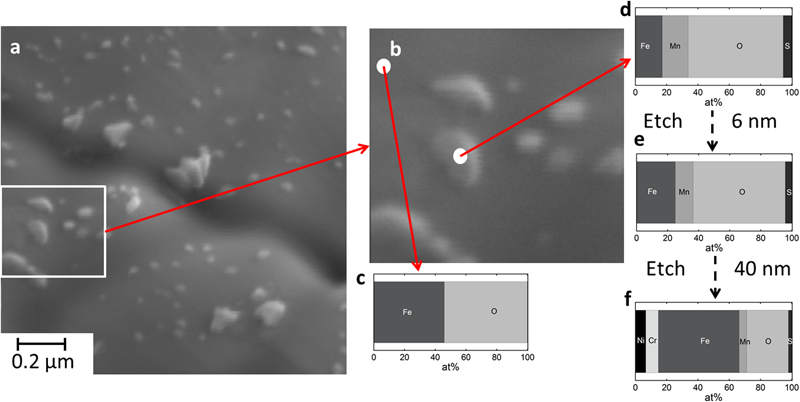
Image (SEM) of a 316L GA<45 μm particle surface, b magnified investigated area, c, d atomic concentrations as determined by AES point analysis, and e, f after Ar+ etching 6 and 40 nm into particles respectively
Table 1 shows the bulk and surface elemental composition of the gas and water atomised 316L powders based on supplier information and XPS surface analysis. No accurate interpretation of Fe(II) and Fe(III) oxidised species was possible due to overlapping binding energies. In accordance with previous findings,2,3 oxidised Si (most probably present as silicates28) was strongly enriched at the surface on the water atomised particles, similar to oxidised Mn [indicative of Mn(III) and Mn(IV) species, possibly MnO2]29 in the case of the gas atomised (GA<4 μm) powder. In both cases, these enrichments increased with reduced particle size.4 There was a significant increase in the binding energy and peak width of oxidised chromium for the WA powder compared to the GA powders [Cr(III) species only] (Table 1), which suggests a possible higher oxidation state. However, due to overlapping peaks between Cr(VI) and Cr(III) species, any accurate assignment was not possible. More speciation sensitive techniques are required.
In order to obtain more detailed information on the oxidation states of the surface oxide metals, cyclic voltammetry using the CPE was applied (Fig. 3). The first cathodic peak (C1) is assigned to adsorbed oxygen.14 Tetravalent manganese was clearly observed in the case of the GA<4 powder, with a peak at −0·41VHg/HgO (C3), and a corresponding anodic peak at −0·11 V (A5), as previously described.14,25 The GA<45 μm powder revealed slightly shifted peaks occurring at −0·44 V and −0·01 V (C4/A6) respectively, which indicate the presence of another Mn containing phase more difficult to reduce. The peak shift may be caused by the Mn rich oxide nanoparticles which are surrounded by (protective) iron oxide (Fig. 2). Trivalent iron was undoubtedly present on all powders, as judged from the cathodic peak at −1·12 V (C9), a corresponding anodic peak at about −0·9 V (A1), and possibly also the small peaks (GA<4 μm powders) at −0·78 V (A2) and about −0·7 V.14,25,30 The oxidation of trivalent to hexavalent chromium was most probably reflected in the strong anodic peak at 0·2 V (A7),31 a peak observed on all powders except the WA<45 μm powder.
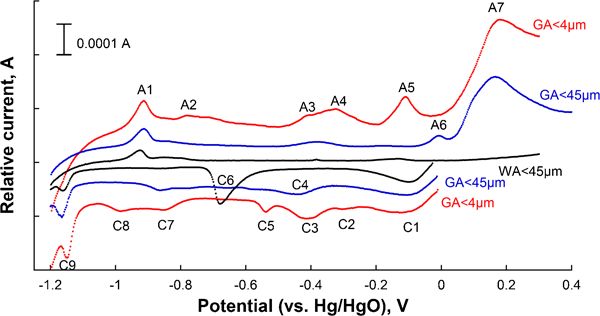
Cyclic voltammograms of inert gas atomised (GA) and water atomised (WA) 316L powders exposed to 8M NaOH (0·5 mV s−1)
Several manganese oxides of tri- or tetravalent oxidation states were observed and assigned for the GA<4 μm powder based on literature data14 including β-MnO2 with cathodic peaks at −0·54 V (C5) and −0·98 V (C8), and corresponding anodic peaks at −0·33 and −0·11 V (A4 and A5).14 In addition to β-MnO2, either Mn3O4 (C8, A4 and A5),14 or Mn2O3 (C3, C8, A4 and A5),14 or both were present. The presence of γ-MnO2 and MnOOH is not possible based on literature data.14,32 The distinct peak C6 (−0·68 V) observed for the WA<45 μm powder (not for the GA powders) was not reflected in any anodic peaks, which indicates that the reduction product of that phase is not oxidised at given conditions. Considering the oxidation/reduction possibilities of surface species at pH 13·1–13·3, only the presence of hexavalent Cr, reduced to Cr(OH)4− (aq) remains as a possible explanation. This observation and the complete absence of the peak corresponding to trivalent chromium oxide (A7) suggest that chromium was predominantly present as hexavalent chromium in the oxide of the WA<45 μm powder. This observation is furthermore supported by higher XPS binding energies for the Cr peak of the WA<45 μm powder compared to the GA powders lacking this oxidation peak. The estimated total transformed mass based on the peak area of C6, given that the peak would reflect a three electrons process as the case for Cr(VI)→Cr(III), was 3·8 μg. This number is compared with a total present oxide mass of 8–25 μg (of which approximately half corresponds to metal components, based on XPS quantification, data not shown) when considering the total surface area of the exposed powder for CV measurements (10 cm2, based on BET area and the investigated mass), the assumption of an oxide thickness of approximately 2–5 nm and an oxide density of 4–5 g cm−3. With a chromium content of 17 wt-% in the surface oxide (relative metal fraction), as determined by XPS, this implies an oxide thickness of 5 nm or more and/or an oxide density exceeding 4 g cm−3 for the WA<45 powder. A one electron transfer, as possible for Fe(III)→Fe(II), Mn(IV)→Mn(III), or Mn(III)→Mn(II), seems hence not possible for the C6 peak, not only due to its potential, but also since the peak area in these cases would correspond to a total mass of 11·4 μg, which at given conditions would correspond to ∼100% of the total mass of the exposed powder.
Raman spectroscopy was employed in order to obtain further information on possible phases (Fig. 4). Hematite, α-Fe2O3, was identified in the case of the GA powder with bands located at 227, 293, 411, 493, 612 and 1300 cm−1.33 The broad band centred at ∼670 cm−1 was not unambiguously assigned as several different possible oxides can be attributed to this band, i.e. Fe3O4, Mn3O4, that reveal a single peak at this band, or MnO2, MnCr2O4, FeCr2O4, MnFe2O4 and δ-FeOOH, that show several peaks.33– 38 In addition, a weak band was for some area measurements observed for the GA powders at 550 cm−1 corresponding to Cr2O3,39 indicating sample heterogeneity. This band was not observed for the WA<45 μm powder in agreement with the CV measurements and indicative by XPS findings. No bands assigned to α-Fe2O3 were observed for the WA powder, with only a single broad band present at 670–680 cm−1. Owing to the lack of other peaks, Fe2O3, Cr2O3 and spinels of the type XCr2O4, where X is Fe or Mn, as well as Fe2SiO4 and chromates, can therefore be excluded for the WA powder.34,40– 42 According to literature findings, this single broad band may be assigned to a magnetite spinel phase (type Fe3O4 or Mn3O4),34,43 or possibly to the silicon bridging oxygen silicon symmetric stretching mode vibration of the pyroxene chain as reported for different silicates, e.g. low iron actinolite.44 This seems most reasonable as oxidised silicon was present to a large extent on the WA<45 μm powder based on XPS findings.
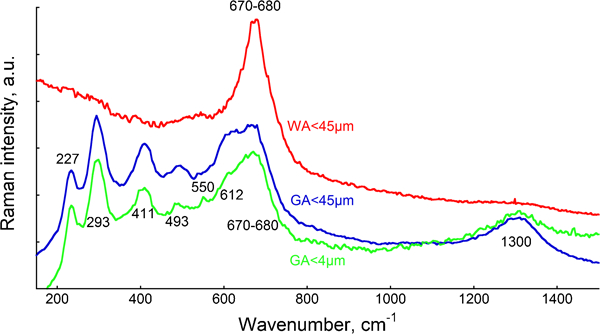
Raman spectra (mean spectra of 2–5 replicate measurements for each powder) determined for GA and WA 316L powders: spectra are off-set for clarity
To the best of the knowledge of the authors, the presence of tri- or tetravalent manganese with oxidising properties16,45 in the surface oxide of the inert gas atomised 316L powder, and the presence of hexavalent chromium within silicate in the surface oxide of the water atomised 316L powder, has not previously been reported. From thermodynamic considerations, tetravalent manganese would not be expected to form at temperatures exceeding 500°C and at low oxygen partial pressures (Fig. 5).
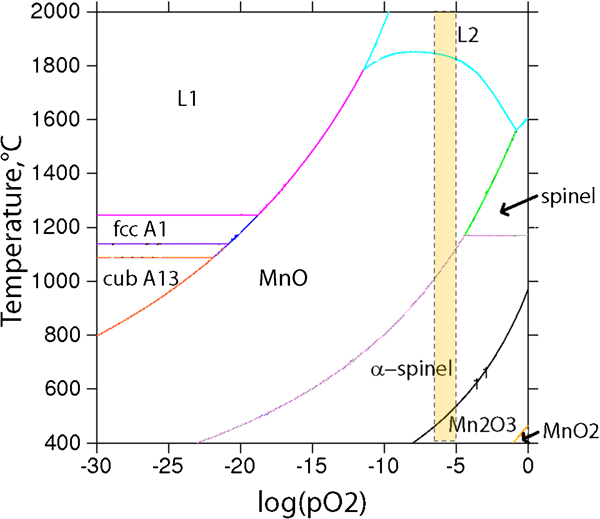
The authors therefore propose that its formation is of kinetic nature, as clearly MnO2 and higher amounts of oxidised manganese were present and identified on the smaller sized, and hence faster cooled, GA<4 μm powder compared with the larger sized powder (GA<45 μm). In contrast to the inert gas atomised powder with only trivalent chromium in the surface oxide, chromium seems to be primarily present in its hexavalent state within the surface oxide of the water atomised powder. Previous findings by some of the authors have shown that the chromium release occurs at very low levels from the WA<45 μm powder after exposure in aqueous solutions and in amounts of the same range as for the GA powders,46 which is in agreement that the oxidised chromium of the WA<45 μm powder is not present as chromate (Fig. 4), which is relatively high soluble. The presence of hexavalent chromium is theoretically possible within a spinel phase, as was reported for the spinel of LiMn2O4.47 Moreover, it has been shown that hexavalent chromium can occur in gaseous form at high (>600°C) temperatures, but only in the presence of water vapour, not in dry gas.48 This may explain the difference in chromium speciation between the gas and water atomised powders. The AES results imply for the GA<45 μm powder that oxidised Mn is present in oxide nanoparticles together with Fe and some amounts of S, a fact that earlier has been suggested to different extent,6,7,26 now experimentally confirmed in this study. However, further in-depth characterisation studies are essential, especially for the water atomised powders, to verify the findings of this study and understand underlying mechanisms.
This study has shown how the oxide composition, morphology and speciation differ between the powders. These findings can assist in a fundamental understanding of solidification processes and surface reactions during atomisation of stainless steel powders. Unfortunately, no detailed information about the atomisation parameters was available. The surface oxide speciation highlighted in this paper may not be important for any further processing, unless the presence of manganese and chromium of higher valent states with possible catalytic properties may be important at given process conditions. Knowledge on surface speciation is however important during occupational exposure and handling of the powders, as it may influence their potential toxic effects. These effects have been17,22,49 and are currently investigated by some of the authors. The dissimilar surface oxide morphology and distribution of the GA<45 μm compared with GA<4 μm powder particles may be important for considerations of micro metal injection molding.
Conclusions
The main aim of this study was to characterise the speciation of surface oxide constituents of inert gas and water atomised stainless steel AISI 316L powders by using Raman spectroscopy, Auger electron and X-ray photoelectron spectroscopy (AES and XPS), scanning electron microscopy (SEM), and cyclic voltammetry measurements.
Oxidised Mn was strongly enriched in the outermost surface oxide of the inert gas atomised (GA) powders, an effect increasing with reduced particle size [Mnox/(Mnox+Feox+Crox) mass fraction: <4 μm (∼47 wt-%), <45 μm (∼13 wt-%)]. Oxidised silicon was strongly enriched [Siox/(Siox+Feox+Crox) mass fraction: 59wt-%] on the surface of the water atomised (WA) powder (<45 μm).
Mn was predominantly present in its tetravalent oxidation state in the gas atomised powders as determined via cyclic voltammetry measurements and indicated by XPS. As investigated for the GA<45 μm particles by means of SEM and AES, Mn was enriched within oxide nanoparticles (20–200 nm) in addition to some Fe and small but enriched amounts of S, compared to an Fe rich matrix oxide, probably Fe2O3.
Cyclic voltammetry, Raman spectroscopy, and XPS binding energy findings imply chromium in the surface oxide of the water atomised powder to be present in its hexavalent oxidation state (not as chromate) and incorporated in a spinel-like and/or silicate phase predominantly composed of Si and to some extent of Fe. These findings are different from observations of the gas atomised powders with chromium in the surface oxide determined in its trivalent oxidation state.
It was clearly shown that the surface oxide composition and speciation of differently sized inert gas atomised and water atomised powder particles are unique and strongly connected to the atomisation procedure. Further studies are needed to understand the influence of different atomisation techniques on the speciation of the surface oxides on stainless steel powders.
Footnotes
Acknowledgements
Experimental help from Oskar Karlsson generating SEM images [Unit for metallic microstructure characterisation (MEMIKA), a joint facility between KTH and Swerea-KIMAB, Stockholm, Sweden], financial support by Cusanuswerk, Germany (for Yolanda Hedberg), and theoretical calculations by Johan Bratberg (Thermo-Calc assessment, Stockholm, Sweden) and invaluable discussions with Dr Olle Grinder are highly acknowledged. Yolanda Hedberg and Inger Odnevall Wallinder are members of the Stockholm Particle Group, an operative network between three universities in Stockholm: Karolinska Institutet, Royal Institute of Technology and Stockholm University, supported by the Swedish Research Councils VR and Formas.