
Abstract
The purpose of this research was to synthesise Fe3O4/SiO2 nanoparticles (NPs) modified by amine groups for bioengineering applications. Magnetic iron oxide NPs were prepared via coprecipitation. The NPs were then modified with a thin layer of amorphous silica, as described by Stober. The particle surface was then terminated with amine groups. The results showed that smaller particles can be synthesised by decreasing the NaOH concentration, which, in the present case, corresponded to 35 nm using 0·9M NaOH at 750 rev min−1 with a specific surface area of 41 m2 g−1. For uncoated Fe3O4 NPs, the results showed an octahedral geometry with saturation magnetisation range of 80–100 emu g−1 and coercivity of 80–120 Oe for particles between 35 and 96 nm respectively. The Fe3O4/SiO2 NPs with 50 nm particle size demonstrated a magnetisation value of 30 emu g−1. The stable magnetic fluid contained well dispersed Fe3O4/SiO2/3-aminopropyltriethoxysilane NPs, which indicated fast magnetic response.
Introduction
The combination of molecular biology and materials science is leading to the development of new devices that benefit from both the exquisite specificity of biomolecules and the extreme control that can be exerted on the properties of the materials, especially of their surface.1 – 7 Studies of iron based materials have attracted much interest in the last few years from both fundamental and applicative points of view.8 – 13 Surface modification of magnetic nanoparticles (NPs) is a frequently used method to promote the performance of NPs as nanobiomaterials. In the recent decades, magnetic NPs, especially Fe3O4 and γ-Fe2O3, have attracted increasing interest because of their outstanding properties, including superparamagnetism and low toxicity and, as a result, their potential applications in various fields, especially in biotechnology and biomedicine, such as cell sorting, enzyme immobilisation, biosensing and bioelectrocatalysis, separation and purification.14 – 17 Various approaches have been explored for the synthesis of high quality magnetic iron oxide NPs.18 – 23 Silica is often employed as the coating material over the surface of iron oxide NPs.24 – 28 Sol–gel process29 – 32 can be used for coating the magnetic NPs with amorphous silica.33 The non-magnetic silica shell is chemically inert, and it can weaken the particle–particle magnetic bipolar interaction and prevent them from agglomeration. Another advantage is that the surface of the coated silica is often terminated by a silanol (Si–OH) group. For the biomedical applications of silica coated magnetic NPs, biomolecules have to be first immobilised on its surface through functional groups deposited on it. Silica represents a material which can be successfully modified for environmental34 and biomedical applications.35 The modification results in products with new functional groups on their surface, capable of interacting with an organic medium, and hence allowing the surface properties to change from typically hydrophilic [silanol groups, which can be easily coupled with 3-aminopropyltriethoxysilane (APTS) by the formation of Si–O–Si covalent bonds36] to hydrophobic (lyophilic) ones.37 The active amino groups (–NH2) facilitate the further functionalisation and can covalently bond with other active groups, such as the carboxyl (–COOH) that can conveniently conjugate with antibodies and other functional groups. The application of amino silane for the modification results in its functional amine group possibly reacting with the carbonyl groups of aldehydes, ketons, esters, amine groups as well as with halides, multiple bonds C = C, epoxy bridges, etc.38 Consequently, specific targeting and multifunctionalisation can be realised. Additionally, the modified magnetic NPs with amino silane shell are non-toxic, biocompatible and injectable.36 This paper deals with the functionalisation and characterisation of magnetite based silica surfaces for biomedical applications, which is based on the deposition of APTS as a precursor.
Experimental
Chemicals
All of the analytic reagents were purchased from the indicated suppliers and used without further purification. Ferric chloride hexahydrate (FeCl3.6H2O, 99%), ferrous chloride tetrahydrate (FeCl2.4H2O, 99%), sodium hydroxide (NaOH, 99%), hydrochloric acid (HCl, 37%), trisodium citrate (TSC), absolute ethanol, ammonia aqueous (25 wt-%) and Si(OC2H5)4 [tetraethyl orthosilicate (TEOS)] were purchased from Merck. 3-aminopropyltriethoxysilane was obtained from Sigma-Aldrich (St Louis, MO, USA). Milli-Q water (specific conductance: 0·1 μS cm−1) was deoxygenated by bubbling N2 gas for 1 h before the use.
Synthesis of Fe3O4
Stock solutions of 1·28M FeCl3.6H2O, 0·64M FeCl2.4H2O and 0·4M HCl were prepared as a source of iron by dissolving the respective chemicals in Milli-Q water under vigorous stirring. In the same way, stock solutions of 0·9–1·5M NaOH were prepared as the alkali sources, and the synthesised Fe3O4 samples were classified as S1–S4, where each sample was synthesised using different NaOH concentrations, i.e. 0·9, 1·1, 1·3 and 1·5M NaOH at 750 rev min−1 correspond to S1, S2, S3 and S4 respectively. Aqueous dispersion of magnetite NPs was prepared by alkalinising an aqueous mixture of ferric and ferrous salts with NaOH at room temperature. Iron source (25 mL) was added dropwise into 250 mL of alkali source under vigorous magnetic stirring (450 and 750 rev min−1) for 30 min at ambient temperature. The chemical reaction of Fe3O4 precipitation is expected as follows
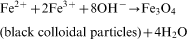
Coating citrate modified Fe3O4 with silica
Following the Stober method, with some modifications, the coating of the citrate modified Fe3O4 with silica was carried out with a basic ethanol–water mixture at room temperature using the obtained magnetite dispersion (only sample S1 at 750 rev min−1 as an optimised value) as seeds. Magnetite dispersion (2 g) was first diluted with water (40 mL) and ethanol (120 mL), and then concentrated aqueous ammonia (3 mL) was added, as suggested in Ref. 33. The resultant dispersion was well dispersed by ultrasonic vibration for 15 min. Finally, 0·9 g of TEOS diluted in ethanol (20 mL) was added dropwise to this dispersion under continuous mechanical stirring. Finally, under continuous mechanical stirring, 0·9 g of TEOS diluted in ethanol (20 mL) was added to the dispersion. After stirring for 12 h, the obtained magnetic product was collected by magnetic separation and washed twice with ethanol. Subsequently, Fe3O4/SiO2 NPs were obtained through the sol–gel approach. For the TEOS concentration used in the present case, the induction period was ∼30 min.
Functionalisation of silica NP surfaces with APTS
As a conventional silanisation reagent, APTS can introduce amine groups onto the surface of the silica nanoshells. In this way, one of the three ethoxy groups (–O–CH2–CH2–CH3) initially present in the APTS molecule breaks off from the molecule, which leads to covalent bonding between the APTS groups and the magnetic NPs. At pH<10, protonation of the amines may give a positive surface charge, which accounts for the high stability of these dispersions. According to Krysztafkiewicz and Binkowski,26 the silica surface, modified with, for examplem APTS, may be presented in Fig. 1.
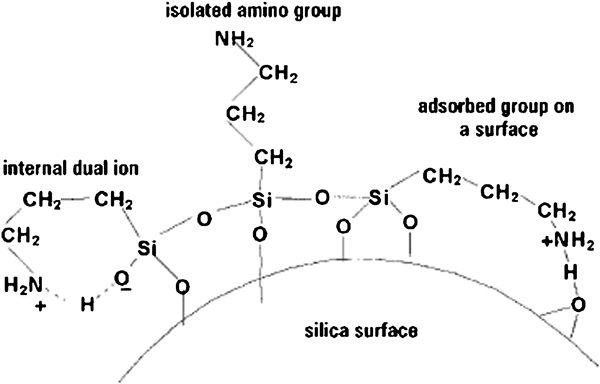
Groups on silica surface following modification with APTS
Silanol (Si–OH) groups are the predominant functional groups at the surface of unmodified silica nanoshells. The amount of APTS solution needed for surface functionalisation is estimated according to the approximate concentration and surface area of the silica nanoshells.39 Consequently, the authors added an excess of APTS (65 μL) to 200 mL of the magnetic NP solution, and the mixture has been vigorously stirred for 2 h. The feasibility of functionalisation reaction could be confirmed visually by observing the precipitation of APTS functionalised magnetic NPs while remaining a clear ethanolic solution at the top. After that, the corresponding covalent bonding was enhanced by refluxing the solution for 1 h. The magnetic NPs were centrifuged at 10 000 rev min−1 and redispersed in 200 mL of ethanol for future use. Amine groups were chosen as the functional group because they are present in proteins, and their chemical interaction with other functional groups is well characterised. In addition, the authors believe that amino modified Fe3O4/SiO2 NPs can be prepared at low cost and, on the other hand, offer a variety of applications.
Measurements
Fourier transform infrared (FTIR) spectra were recorded by an EQUINOX 55 FTIR spectrometer. X-ray diffraction (XRD) measurements were performed at room temperature using a FK60-04 X-ray diffractometer with Cu radiation. Thermogravimetric analysis (TGA) was carried out using a TGA-PL analyser (England) from 25 to 650°C under the nitrogen atmosphere with a heating rate of 5°C min−1. Before the measurements, the samples were degassed at 160°C in a vacuum for 6 h. The Brunauer–Emmett–Teller (BET) method was utilised to calculate the specific surface areas by a Quantachrome TPR Win v1·0 using nitrogen as the sorbate. Scanning electronic microscopy (SEM) images were recorded by a Philips XL30 electron microscope (The Netherlands) at an accelerating voltage of 30 kV. Transmission electron microscopy (TEM) was performed using a Phillips CM-200-FEG microscope operating at 120 kV. Magnetisation measurements were carried at 300 K in a magnetic field H of up to 20 kOe with a vibrating sample magnetometer (PAR 155) that can measure magnetic moments as low as 10−3 emu. For the magnetisation measurements, uncoated Fe3O4 NPs were in dry powder form obtained by evaporating the water from the solution.
Results and discussion
It is worth mentioning that flowing N2 gas not only protects the critical oxidation but also reduces the particle size. A complete precipitation of Fe3O4 should be expected between 7·5 and 14 pH while maintaining a molar ratio of Fe2+/Fe3+ = 1∶2 under a non-oxidising environment; otherwise, Fe3O4 might also be oxidised. An octahedral Fe3O4 with high quality and crystallinity could be obtained in the concentrated solution. As shown in Fig. 2, the mean Fe3O4 particle size examined by TEM imaging exhibited an octahedral-like geometry with an almost dispersed state. The powder size measurement showed a gradual increasing trend with increasing percentage of NaOH, which, in the present case, corresponded to 35 nm at 0·9M NaOH (S1) to 100 nm at 1·5M NaOH (S4) at 750 rev min−1 (Fig. 3). The reason may be due to the reaction mechanism of magnetite
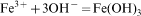
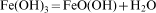
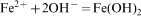
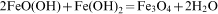

Image (TEM) of uncoated Fe3O4 NPs (samples S1 and S4) at 750 rev min−1
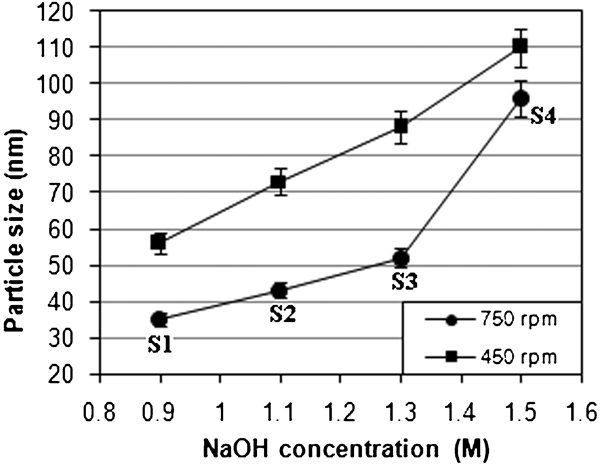
Particle size versus NaOH concentration for uncoated Fe3O4 NPs at different revolutions per minute values
Saturation magnetisation Ms, residual magnetization Mr, coercivity Hc and bulk saturation Mb = ρMs are among the main magnetic parameters that have to be characterised when considering the magnetism of a magnetic material. The value of Ms for these particles was measured to be ∼82 emu g−1, which steadily increased by increasing their size (Fig. 4). It can be seen that the saturation magnetisation of Fe3O4 NPs is close to the bulk value of magnetite, which is about 85–100 emu g−1.19 One new result found in the present experiment is the relatively higher magnetisation saturation of Fe3O4 (82 emu g−1) compared with usual values.33 In addition, the corresponding values of Hc were measured and plotted in Fig. 5 as 119 and 82 Oe for particles sizes of 35 and 96 nm respectively.
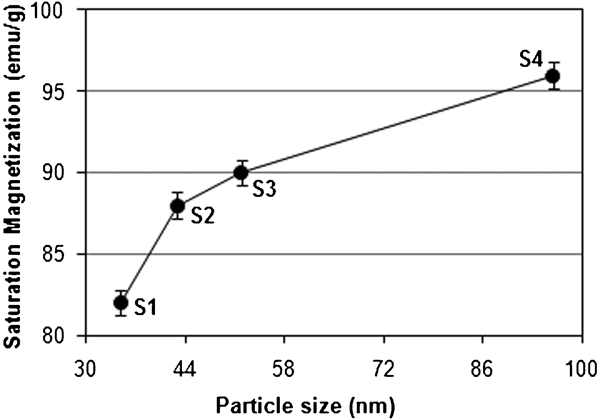
Variation of saturation magnetisation with particle size for uncoated Fe3O4 NPs at 750 rev min−1
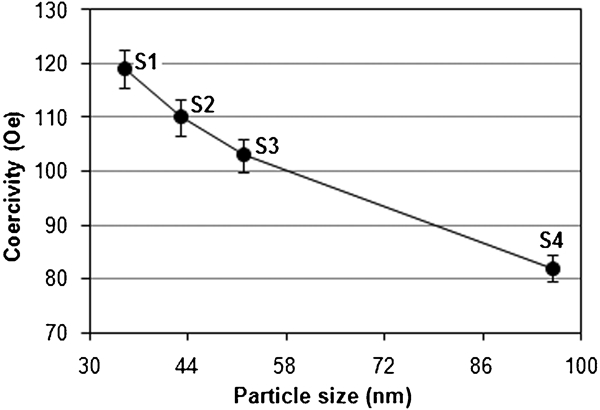
Variation of coercivity with particle size for uncoated Fe3O4 NPs at 750 rev min−1
These results suggest that Hc is strongly size dependent, and bulk samples with sizes larger than the domain wall width can cause magnetisation reversal due to domain wall motion. As domain walls move through a sample, they can become pinned at the grain boundaries, and additional energy is needed for them to continue moving. Pinning is one of the main sources of Hc. The grain size dependence of Hc and the permeability theory40 predict
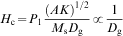
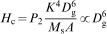
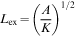
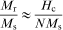
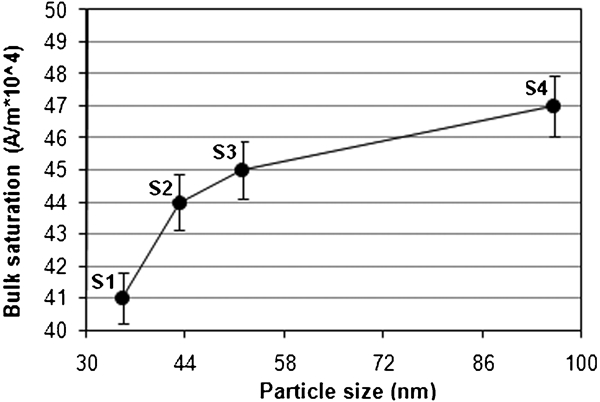
Variation in bulk saturation with particle size for uncoated Fe3O4 NPs at 750 rev min−1
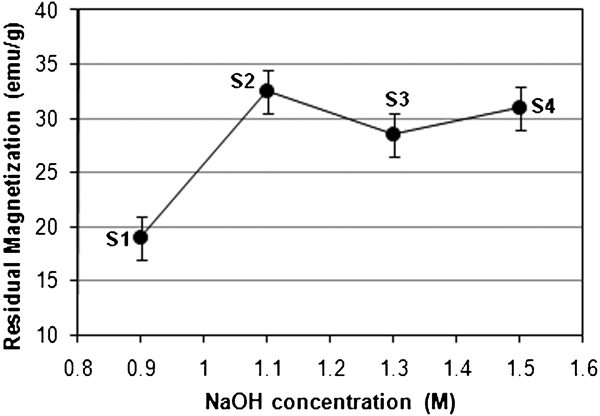
Variation in residual magnetisation with particle size for uncoated Fe3O4 NPs at 750 rev min−1
It is worth mentioning that SQ characterises the shape of the hysteresis loop. A perfect hysteresis loop has an SQ of 1. The SQ ( = Mr/Ms has an increasing trend as the applied field is increased and that the SQ for the perpendicular configuration is higher than the that for the parallel one.44 The non-linear variation of residual magnetisation of particles with their size can be attributed to the following: first, there is a non-uniform distribution of particle size within a given sample group (e.g. S1–S4), which implies a random selection of a particle for TEM purpose; second, the particles are randomly oriented without the previous magnetic orientation treatment; and third, some magnetite can be changed into maghemite (γ-Fe2O3) due to oxidisation.
The magnetic NPs were easily coated with amorphous silica. The reaction mechanism is explained as below
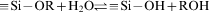

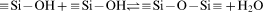
The SiO2 shell allows each Fe3O4 particle to behave independently, and interparticle interactions are therefore not important. Figure 8 shows the room temperature magnetisation curve of the Fe3O4/SiO2 NPs obtained using a vibrating sample magnetometer. The M (H) hysteresis loop for the Fe3O4/SiO2 NPs was almost completely reversible. It means that the magnetisation curve exhibits zero remanence and coercivity, which proves that Fe3O4/SiO2 NPs has superparamagnetic properties. The Ms value for the Fe3O4/SiO2 nanostructures was 30 emu g−1 at an applied magnetic field of 6000 Oe, which is ∼3·6% of the Ms for Fe3O4.
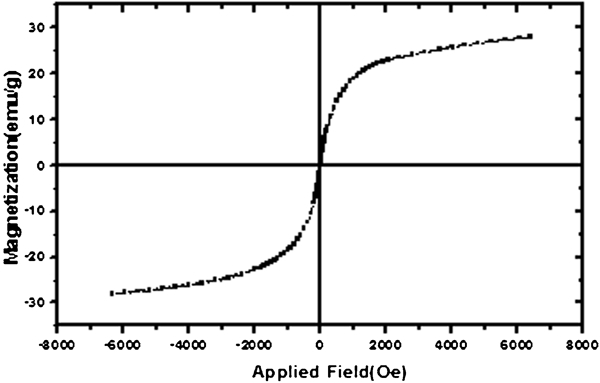
Variation in Fe3O4/SiO2 NP magnetisation with applied field
When the external magnetic field was removed, the core–shell particles could redisperse rapidly, which is an advantage to their bioengineering applications.
Figure 9 shows the XRD patterns for uncoated Fe3O4 NPs, citrate modified Fe3O4 NPs and SiO2 coated Fe3O4 NPs, with each pattern normalised to its maximum intensity. The peaks are indexed with the fcc structure corresponding to magnetite phase. The average size of the crystals was estimated using Scherrer's formula
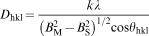
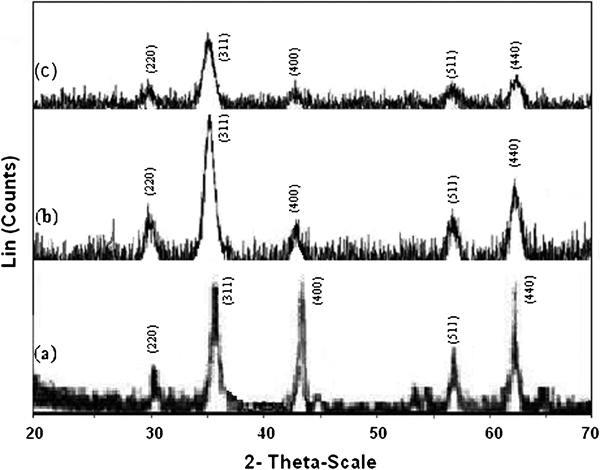
X-ray diffraction patterns for a uncoated Fe3O4 NPs, b citrate modified Fe3O4 NPs and c SiO2 coated Fe3O4 NPs: all indicated that Miller indices in a–c correspond to magnetite
Figure 10 indicates the results of the SEM size analysis of magnetic NPs, where a mean diameter of 30 nm was determined. As shown in Fig. 10b, the spherical core–shell products have a rough surface. The silica layer can stabilise the magnetic particles in two different ways: one is by sheltering the magnetic dipole interaction, and the other is by bringing negative charges on the surface of shells, which enhances the coulomb repulsion of the magnetic particles. Meanwhile, upon the increase in particle size, their morphologies become more regular in spherical shape due to the requirement of minimum surface energy. As discussed by Pham et al.,39 there is no discernible difference in size for the purified NPs between pre- and post-functionalisation with APTS. To observe the particle size distribution of Fe3O4/SiO2 NPs, TEM was used (Fig. 11). A typical size of the core–shell structure was measured at ∼50 nm, which is comparable with the results obtained by Deng et al.24 The uniform dispersion of functionalised NPs would make them a suitable candidate for bioengineering applications.

Images (SEM) of a citrate modified Fe3O4 NPs and b SiO2 coated Fe3O4 NPs

Image (TEM) of Fe3O4/SiO2 NPs
Infrared spectroscopy can provide some useful information regarding the surface structure of NPs, mainly about the order and packing of the surface chains. The main absorption peaks of around 591 and 3422 cm−1 in Fig. 12a correspond to Fe–O and O–H stretching vibration modes respectively. In addition, the new absorption peaks at 1614 and 1391 cm−1, which appear in the FTIR spectra of the citrate modified Fe3O4 NPs, are characteristics of the carboxylate bonds. The presence of magnetite is evident at 418 and 462 cm−1. The absorption peaks of the citrate modified samples at 1622 and 1397 cm−1 are due to the COO–Fe bond (Fig. 12b). The characteristic absorption for the silica network is assigned as follows. The broad high intensity band at 1092 cm−1 is due to the asymmetric stretching bonds of Si–O–Si in SiO4 tetrahedron associated with the motion of oxygen in Si–O–Si antisymmetric stretch. The band at 802 cm−1 is assigned to the Si–O–Si symmetric stretch. The band at 466 cm−1 is an indication of the presence of Si–O–Fe (Fig. 12c). Figure 10d shows the IR spectra of APTS coated Fe3O4 NPs. The broad bands near 1092 and 798 cm−1 are the contribution of Si–O. The adsorption bands in 2930 and 2850 cm−1 are also due to the stretching vibration of –CH2, and those near 3430 and 1633 cm−1 are assigned to –NH2.
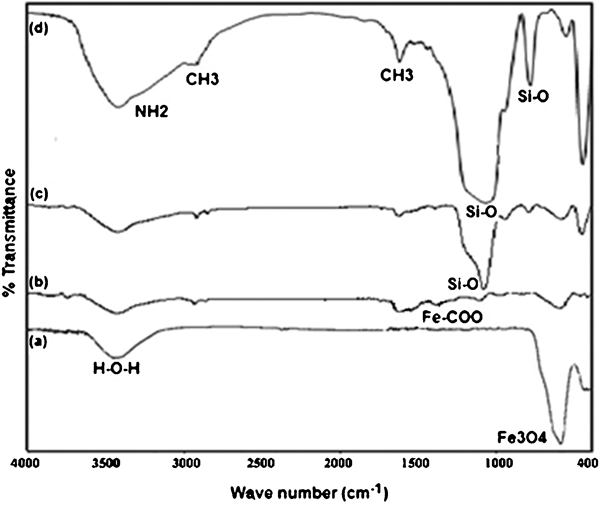
Spectra (FTIR) of a uncoated Fe3O4 NPs, b citrate modified Fe3O4 NPs, c SiO2 coated Fe3O4 NPs and d Fe3O4/SiO2/APTS NPs
Each dried sample (0·01–0·02 g), after being accurately weighted, was placed in a sample tube. The surface area of all the samples was determined by the BET method. Figure 13 shows the NaOH concentration versus the specific surface area for Fe3O4 NPs. The N2 adsorption–desorption isotherms demonstrated that the highest specific surface area of 41 m2 g−1 was achieved at 750 rev min−1 using 0·9M NaOH. Using the BET values, one can calculate the amount of particle size DBET considering the following equation
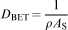
Specific surface area versus particle size for uncoated Fe3O4 NPs at 750 rev min−1
Brunauer–Emmett–Teller (BET) measurements

The heat endurance of Fe3O4/SiO2/APTS NPs was evaluated by thermogravimetric test (Fig. 14). The Fe3O4/SiO2 nanoshell indicated three distinct weight loss stages
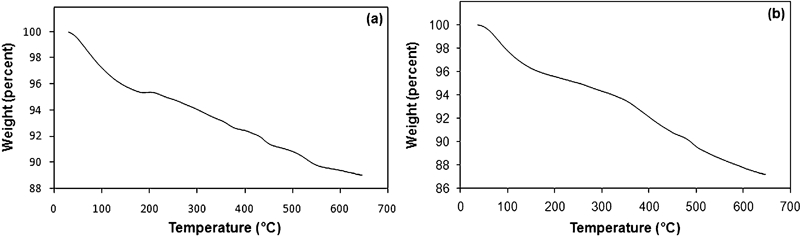
Graphs (TGA) of a Fe3O4/SiO2 NPs and b Fe3O4/SiO2/APTS NPs
a small weight loss in the range of 40–180°C mainly due to the evaporation of residual alcohol, the physically adsorbed water and the slight dehydration of silanol groups
a large weight loss in the range of 200–450°C, which is attributed to the decomposition of organic substances
a minor weight loss at the higher temperatures of 500–550°C due to the complete dehydration of silica species, which thereafter remain to be stable.
According to TGA analysis, the residual mass per cent of silica coated NPs is 89% at 550°C, while that of SiO2–NH2 is 87%, which indicates that the grafting percentage of APTS is ∼6%.
Conclusions
In conclusion, the authors have presented a controlled coprecipitation method which demonstrates the feasibility of synthesising magnetite NPs. For uncoated Fe3O4 NPs, the results showed an octahedral geometry with high saturation magnetisation. Highly stable magnetic fluid containing well dispersed citrate modified magnetite NPs was obtained. It was shown that the surface of SiO2 coated Fe3O4 NPs synthesised by coprecipitation and Stober methods can be modified biologically for biomedical applications. This synthesis provides a rapid and effective route to magnetic core–shell particles that are soluble in aqueous media. During the hydrolysis and condensation of TEOS, the formed primary particles are effective in suppressing the dipole–dipole interactions among the magnetic particles and hence providing a better prevention from agglomeration compared with Fe3O4 NPs alone. The SiO2 shell results in the decrease in the magnetic strength of the core–shell NPs due to the weight contribution from the non-magnetic SiO2. Based on the FTIR information, the authors conclude that Fe3O4 is covered by TSC, TEOS and APTS layers that exhibit their characteristic IR vibration bands. The XRD results confirm that the iron oxide NPs before and after coating have the Fe3O4 phase. These results provide a better understanding of the structural and magnetic properties for amino modified silica coated Fe3O4 NPs and thus are very valuable for evaluating the future applications of this class of magnetic NPs in bioengineering applications.