
Abstract
Meat and bone meal ash, mixed with recycled soda–lime–silica glass and small amounts of additives, was successfully valorised in the processing of sintered glass ceramics, after melting and forming two calcium phosphate glasses. Sintering was applied to fine powders (<37 μm) at temperatures of 700–1070°C for 0·5–2 h, after very rapid heating (40°C min−1). Mixtures with small additions of CaO and CaF2 led to fluorapatite–wollastonite glass ceramics, which retained a significant porosity even at 1070°C, due to the delay in viscous flow caused by rapid crystallisation. This feature was exploited for strong open celled macrocellular glass ceramics, obtained by sintering glass powders mixed with polyethylene sacrificial templates. Mixtures with small additions of CaO and Na2O led to dense and strong combeite glass ceramics (bending strength, >100 MPa), sinterable at particularly low temperatures (800°C). Both porous and dense glass ceramics could be exploited as low cost and high strength materials, or even as biomaterials, due to the biocompatibility of the crystal phases.
Introduction
In addition to being extensively applied for the safe immobilisation of high level radioactive wastes,1,2 vitrification is now becoming increasingly attractive for the safe treatment of a number of toxic, non‐radioactive wastes.3–5 Vitrification provides substantial volume reduction and the immobilisation of hazardous waste components in a chemically inert glass matrix. However, this approach may not yet be economically viable for the treatment of many non‐radioactive wastes since the vitrification process is capital and energy intensive. This economic difficulty may be overcome if it is considered that waste materials may be used themselves as raw materials in the production of materials for use in many applications. Among the most widely studied examples is the production of glass ceramics from wastes, with a range of potential applications.3,6 Recent studies7–10 have evidenced that waste glasses can be successfully converted into sintered glass ceramics with particularly economical processing, similar to that of ceramic frits. Finely ground glass powders (typically <40 μm, easily obtained by grinding glass not previously subjected to annealing, e.g. glass poured into cold water or onto cold steel plates), undergo sinter–crystallisation, i.e. viscous flow sintering with concurrent crystallisation, at temperatures of 900–1050°C, within very short times (often <1 h) and with high heating rates. In some cases, sintered glass ceramics may be manufactured by ‘direct heating’, i.e. by the introduction of samples directly at the sintering temperature.7–10 This rapid crystallisation generally depends on surface nucleation, which is greatly enhanced for fine glass powders.11
Incineration is now firmly established as a key method of waste disposal and volume reduction, with the added benefit of generating energy from waste. In some cases, incineration is viewed as a hygienic solution: for example, the destruction of organic matter in animal waste can be safely achieved by incineration. Meat and bone meal (MBM) is one such waste, and with increasing restrictions on the reuse of untreated or raw MBM, incineration (or preferably generating energy from waste), followed by disposal, has been widely adopted. The potential reuse of incinerator ashes as raw materials in other processes has received considerable interest owing to its environmental and economic benefits. Research to date has focused primarily on the ashes arising from combustion of coal (power stations) and municipal solid waste (incinerators) owing to the high volumes and ready availability of these ashes. However, MBM ash has thus far received little research interest in this regard, despite its availability in considerable quantities (3·5 Mt MBM was incinerated in the EU in 2007).12 Moreover, because MBM ash arises from the incineration of MBM, it has a chemical composition dissimilar to those of the majority of other incinerator ashes (which include coal ash and municipal solid waste ash and are typically silica rich). Meat and bone meal ash essentially consists of calcium phosphate with various impurities.12,13 While the cement industry has increased its use of MBM as a fuel in kiln firing12 and research has been published into the potential use of MBM ash in cements and mortars, the majority of MBM ash is presently disposed of in landfill when it could instead be used as a raw material.13 Given the high percentage of calcium phosphate present in MBM ash12–14 and also given the cost of calcium phosphate (which can be useful in the production of ceramic and glass ceramic materials), it is important to consider the reuse opportunities of MBM ash in such applications. Recent Japanese research15 has considered the use of incinerator fly ash and chicken bone ash in value added calcium phosphate hydrogel manufacture, highlighting the growing interest in avoiding landfill of calcium phosphate rich wastes and reusing them in value added applications.
In this paper, the authors present a new approach to the valorisation of MBM ash, aimed at producing sintered glass ceramics that could be exploited as low cost and high strength materials or even as biomaterials. Meat and bone meal ash, i.e. a phosphorous rich waste, was mixed with common soda–lime–silica (SLS) glass cullet, i.e. a silica rich waste, and small amounts of secondary additives, and vitrified. Soda–lime–silica glass cullet is a different kind of waste to incinerator ash: although recommended for limiting the consumption of energy and natural raw materials in the production of commercial glass,16 the usage of scrap glass or ‘cullet’ in manufacturing new glass articles is possible only after its separation from metallic and ceramic contaminants. This separation leads to a fraction of almost pure glass ready for the industrial use and a fraction enriched in contaminants, which is practically unemployed and mostly landfilled, or is used in low value applications, such as road building.17 Since the vitrification of MBM ash and SLS glass generally leads to the formation of SiO2–P2O5–CaO materials, in the present research, the authors have focused on compositions leading to valuable glass ceramics from this particular system, i.e. those corresponding to the formation of apatite–wollastonite or sodium calcium silicate (e.g. combeite) upon devitrification, both of which are widely recognised as phases providing optimum biocompatibility.6,18 The target compositions were approached by carefully balancing the contents of MBM ash and SLS glass and by the introduction of Na2O, CaO and/or CaF2 as minor additives. The two formulations considered demonstrated a different densification, owing to a different equilibrium between viscous flow sintering and crystallisation. The mixture based on apatite–wollastonite led to porous glass ceramics, while the mixture based on combeite led to dense glass ceramics (i.e. with a porosity below 3%), exhibiting a remarkable bending strength of more than 100 MPa. Porosity is not a critical issue in a number of applications, including biomaterials, if it is considered that natural bone is well known to be a cellular material.19 Nevertheless, it is useful for most applications, whether porosity is critical or not, that the mechanical properties are optimised to maximise the range of potential applications: here, strong open celled macrocellular apatite–wollastonite glass ceramic foams were obtained by a careful control of the microstructure, in turn associated with the sintering of fine glass powders mixed with polyethylene (PE) sacrificial templates.
Experimental procedure
The chemical composition of MBM ash is reported in Table 1. Two different glasses were obtained by mixing MBM ash with SLS cullet: the first, G1, corresponds to the addition of CaCO3 and CaF2 (reagent grades, >99% purity); the second, G2, refers to the addition of CaCO3 and Na2CO3 (reagent grades, 99% purity). Details about the proportion between wastes and additives are given in Table 1. The formulations of the two glasses are broadly similar to those of Kokubo's Cerabone and Hench's Bioglass (G2).6,20 The authors’ glasses, G1 and G2, were obtained by melting well mixed batches of MBM ash, SLS cullet and additives. Batches to make 300 g glass were placed in recrystallised alumina crucibles and preheated overnight to 1030°C. The crucibles were then transferred to an electrically heated glass melting furnace at 1400°C, and glasses were melted for 3 h before pouring onto cold steel plates. The glasses were subjected to dry ball milling and sieved to powders with a dimension <37 μm. Dilatometric analysis (402E Netzsch Gerätebau GmbH, Selb, Germany) was performed on glass fragments of both compositions, while differential thermal/thermogravimetric analysis (STA409; Netzsch Gerätebau GmbH, Selb, Germany; operating at 10°C min−1 heating rate) was performed on fine glass powders with a dimension <37 μm.
Waste raw materials, nominal chemical composition (wt‐%) and characteristic temperatures of investigated glasses
Sintering experiments were first performed on disc samples with a diameter of 30 mm and a height of 2 mm, obtained by uniaxial pressing at 40 MPa of fine powders in a cylindrical steel die at room temperature, with no added binder. Sample discs were subjected to direct heating, consisting in the insertion of samples into the furnace at the sintering temperature, in the interval 700–1070°C for 0·5–2 h, followed by sudden cooling by removal from the furnace and cooling in air. The bulk density was estimated by applying the Archimedes’ principle on fragments of disc samples. X‐ray diffraction analyses (Bruker D8 Advance, Karlsruhe, Germany) were performed on powdered samples, employing Cu Kα radiation (0·15418 nm), in the interval 15–75° 2θ in steps of 0·05° 2θ with 2 s counting time. The diffraction patterns were analysed using the Match! software package (Crystal Impact GbR, Bonn, Germany), supported by data from PDF‐2 database (International Centre for Diffraction Data, Newtown Square, PA, USA).
Selected sintering treatments were applied on larger samples, with dimensions of 40×30×3 mm, obtained by uniaxial pressing in a rectangular steel die. These samples were sintered with a heating rate of 40°C min−1 and with natural cooling after the holding stage. The large samples were cut into small beams of about 3×2×30 mm for bending strength determination. All sample beams were carefully polished to a 6 μm finish and chamfered at the edges, using abrasive papers and diamond paste. Young's modulus E was measured by non‐destructive resonance frequency testing (GrindoSonic Mk5, Leuven, Belgium). Four‐point bending tests (24 mm outer span and 8 mm inner span) were performed using an Instron 1121 UTS (Instron, Danvers, MA, USA), with a crosshead speed of 1 mm min−1. Each data point represents the average of at least 10 individual tests. Selected polished samples were employed for Vickers indentation tests (DG 901 microindenter; Officine Galileo, Florence, Italy) at low load (10 N), which yielded the microhardness (HV). The bending bars were employed also for density determination by geometrical measurements and by application of the Archimedes’ method. Gas pycnometry (Micromeritics AccuPyc 1330, Norcross, GA, USA) was applied to powdered bars for determination of the true density. The amounts of open and closed porosities were estimated by considering the differences in the measured densities: for a given mass, geometrical measurements refer to the volume comprising open and closed pores, while the volume inferred from water immersion is deprived of open pores; finally, the volume associated with the true density is obviously related only to the solid phase.
Glass G1 was also employed for the preparation of macrocellular foams. Fine glass powders were mixed with PE spheres (diameter, >420 μm range; Clariant Italia SpA, Milan, Italy) in a solution of silicone resin (MK, Wacker‐Chemie GmbH, München, Germany) in isopropyl alcohol, in the following proportions: G1/PE/MK/isopropyl alcohol = 8 g/8 g/1 g/10 cc. The usage of a silicone resin, acting as a binder, was derived from a previous work concerning cellular glass ceramics.21
The obtained slurry was first homogenised by manual shaking in a PE bottle with porcelain balls, then cast into a polystyrene container and dried overnight at room temperature. The dried slurry was manually ground with pestle and mortar, thus producing small agglomerates, which were pressed (at 40 MPa) in the abovementioned cylindrical die.
The obtained PE/glass composites were finally subjected to a multistep thermal treatment. This thermal treatment consisted of two holding stages, each of 2 h, at 220 and 370°C, attained by a slow heating rate (3°C min−1) and aimed at the burnout of PE spheres, according to a recent paper by Lombardi et al.,22 and finally with rapid heating to 1070°C at 40°C min−1, followed by a holding stage of 30 min and natural cooling, aimed at the sinter–crystallisation of glass powders.
The microstructure of all samples was investigated by scanning electron microscopy (JSM‐6490; JEOL, Tokyo, Japan); polished surfaces of dense glass ceramics were analysed after HF etching (3 min in 5%HF).
Results and discussion
Density–crystallisation correlation (tests on disc samples)
Sinter–crystallisation consists of overlapping viscous flow sintering and crystallisation. It must be noted that the two phenomena may contrast with each other: if crystallisation occurs before most of viscous flow sintering is completed, the enhancement of apparent viscosity provided by the crystal inclusions hinders further densification, thus leading to porous materials.23
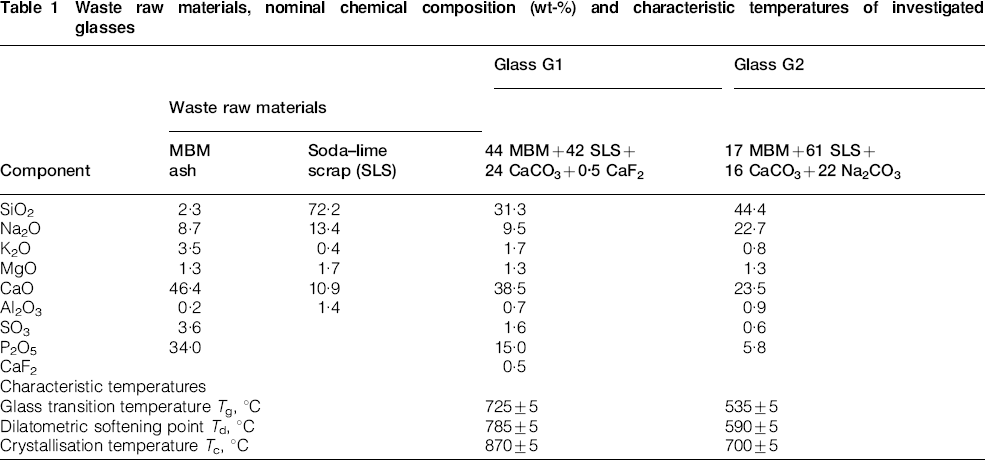
As in previous papers discussing sinter–crystallisation,7–10 the crystallisation temperature Tc of 870°C (see Table 1) was selected as a reference for G1 glass. Sintering was performed at Tc−50°C, Tc, Tc+100°C and Tc+200°C. All samples were evidently porous, as expected on the basis of their relatively high dilatometric softening point Td of ∼790°C (i.e. only 80°C below Tc, indicating the likelihood of insufficient viscous flow to avoid porosity). Crystallisation took place before an intensive viscous flow, which is generally available only with sintering temperatures of about 100–150°C greater than the dilatometric softening point.24 For G1 glass, these temperatures largely exceed Tc, with the likely precipitation of crystals, which delay the densification by greatly increasing the apparent viscosity. As illustrated by the diffraction patterns shown in Fig. 1a, crystallisation effectively occurred even at 820°C.
X‐ray diffraction patterns of glass ceramics from G1 glass
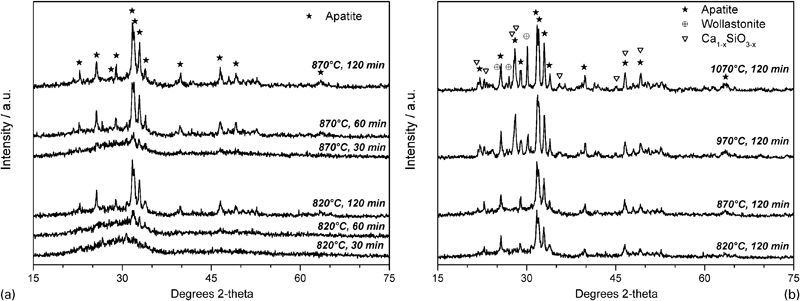
The primary crystalline phase is identified as apatite; the automatic crystal phase detection provided by the Match! program package indicated fluorapatite [Ca5(PO4)3F, corresponding to the PDF card no. 71‐0880] as the most probable phase. In the opinion of the authors, however, a more appropriate phase would be oxyfluorapatite [i.e. Ca10(PO4)3(O,F2)], as reported in the literature,6 but not available in the adopted crystallographic database, since the overall fluorine content of our samples is too limited to support the extensive presence of fluorapatite. The precipitation of this apatite phase has a rapid increase with the sintering time: from Figs. 1a and 2, it may be seen that the intensity of the peaks, well known to be proportional to the quantity of a certain phase, more than doubled for a time increase from 30 to 120 min at 820°C. It may be also observed that increases of sintering temperature catalyse the precipitation: if, in Fig. 2, the level of intensity (referred to the main peak of the apatite phase) associated to 120 min at 820°C is considered, it was almost achieved after 60 min at 870°C and greatly exceeded after only 30 min at 970 and 1070°C. Figures 1b and 2 illustrate that also wollastonite, β‐CaSiO3 (monoclinic, PDF card no. 84‐0655) developed, with significant precipitation mainly at high temperatures. The formation of this secondary phase is consistent with the similarity of G1 composition to that of apatite–wollastonite glass ceramics.6,20 For long treatments (120 min), at 970–1070°C, a further calcium silicate, with the formula Ca1−xSiO3−x (CaO/SiO2 molar ratio below 1; PDF card no. 26‐1069), was detected.
Variations in intensity of main characteristic diffraction peaks of apatite and wollastonite phases in glass ceramics from G1 glass
The lack of adequate crystallographic data for the obtained phases in glass ceramics obtained from glass G1 impeded a semiquantitative analysis by the same Match! software package, but due to the almost flat background of diffraction patterns for long treatments at high temperature, i.e. 120 min at 1070°C (see Fig. 2b), the amount of amorphous phase may be assumed to be small.
Archimedes’ method was applied in order to obtain an approximate but rapid indication of the bulk density, in the presence of geometrically irregular samples (some broke readily into fragments on cooling due to the cracks developed upon pressing with no binder). It is well known that Archimedes’ method is correctly applied for samples with a limited open porosity: for samples from G1 glass, which were evidently porous, the measurements were made quickly after immersion, in order to minimise the filling of open pores and thus obtain a rough indication of the overall densification. The estimated density had a very limited increase, of ∼5%, with increasing sintering time and temperature, passing from 2·67 g cm−3 after 30 min at 820°C to 2·80 g cm−3 after 120 min at 1070°C. These values are quite low, compared to the density of Kokubo's Cerabone (3·07 g cm−3).25
Sinter–crystallisation of glass G2 was more straightforward than that of glass G1. Despite the fact that the crystallisation temperature was only slightly more distant from the dilatometric softening point than in the case of G1 (see Table 1), G2 glass ceramic samples were visibly less porous and demonstrated a brilliant and smooth surface (open porosity was therefore negligible). However, also in this case, the density variations were very limited: at 700°C, the temperature of the crystallisation exothermic peak, the density was ∼98% of the maximum density of all glass ceramics from G2, being 2·73 g cm−3.
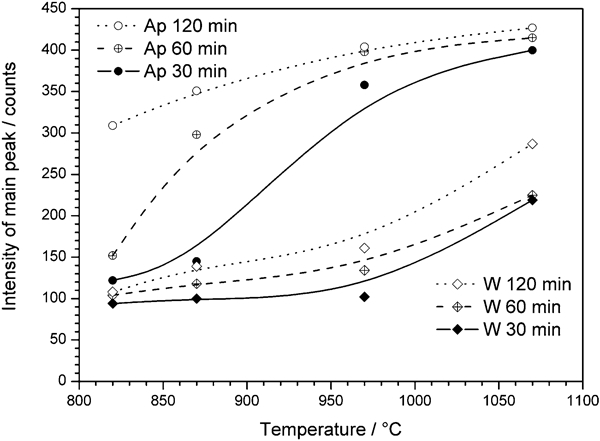
The most significant changes in the thermal treatment of G2, as shown in Fig. 3, are associated with the crystallisation, which increases with the sintering temperature, at a constant holding time of 30 min; a further increase is associated with a longer holding time, i.e. 60 min. Longer treatments at 800°C, up to 120 min, did not cause a significant variation except for the increase in a secondary crystal phase. The dominant crystal phase is identified as combeite (2·2Na2O.3·8CaO.6SiO2, corresponding to the PDF card no. 78‐1649; this corresponds to combeite with a slightly higher Na2O/CaO molar ratio than standard combeite,18 Na2Ca2Si3O9, i.e. Na2O.2CaO.3SiO2). For long treatments at 800°C, the presence of another sodium–calcium silicate phase (identified as Na2O.CaO.SiO2 or Na2CaSiO4, corresponding to the PDF card no. 73‐1726) cannot be excluded. Such a phase would justify the increase in the reflected peak intensity at ∼33° 2θ, associated with both crystal phases, compared to that at ∼34° 2θ, associated only with combeite.
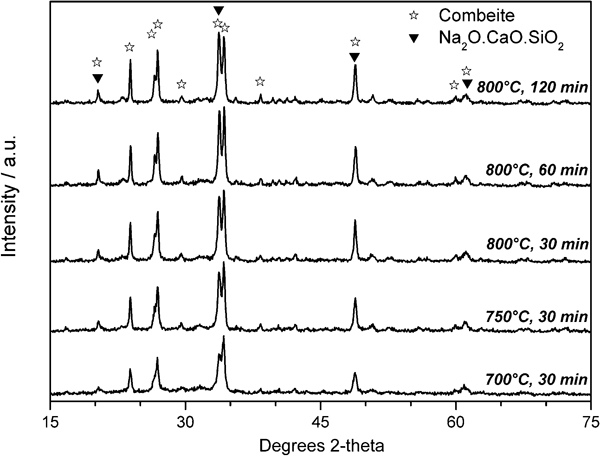
X‐ray diffraction patterns of glass ceramics from G2 glass, sintered in various conditions
The formation of combeite is consistent with expectations since Hench's bioactive glass composition, which is similar to that of G2, is known to most often lead to formation of Na2Ca2Si3O9 crystals upon heat treatment.26 From the semiquantitative analysis provided by the Match! software package, the overall crystallinity at 800°C is ∼80 wt‐% (60% combeite, 20% secondary sodium–calcium silicate); this means that sodium and calcium oxides and silica were largely incorporated by the crystal phase, the residual glass presumably being rich in P2O5. The bioactivity of combeite is widely reported in the literature,26–28 and it is noted that combeite is found to be even more strongly bioactive than fluorapatite.18,29 The most interesting point, in the authors’ opinion, is that crystallisation is obtained at very low temperature and with a very limited processing time, significantly shorter than those employed for the crystallisation of similar compositions by conventional nucleation and crystal growth treatments (see, for example, Abo‐Mosallam et al.,18 who discussed combeite containing glass ceramics crystallised after 5 h at 615–720°C, followed by 10 h at 775–815°C). The sinter–crystallisation approach is therefore confirmed to be extremely effective for relatively low cost processing of glass ceramics.
Characterisation of larger glass ceramic samples and possible applications
The remarkable porosity of small samples from G1 glass is confirmed in the larger samples, sintered at 1070°C for 120 min (the condition that maximised both density and crystallisation): as reported in Table 2, a total porosity of ∼8·6%, with an open porosity of ∼7%, is estimated from density measurements; it is interesting to note that the true density (3·02 g cm−3) is very close to that of Cerabone. The SEM image shown in Fig. 4a confirms the presence of pores.

Microstructures of glass ceramics obtained from G1 glass
Summary of physical and mechanical properties of large glass ceramic samples from G1 and G2 glasses
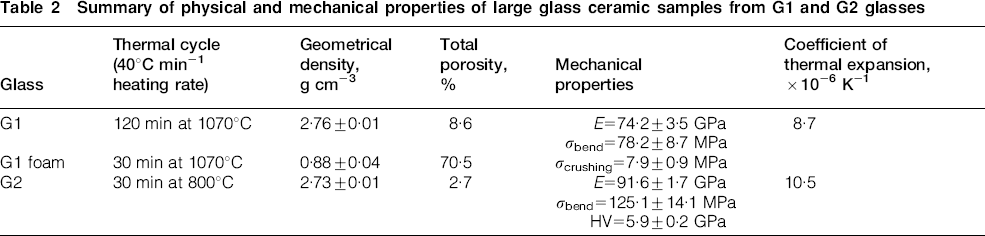
Despite porosity, the glass ceramic from G1 glass possesses interesting mechanical properties: in fact, the values of elastic modulus and bending strength are 74 GPa and 78 MPa respectively, quite close to those of denser glass ceramics obtained for structural applications (e.g. architectural) applications.6
The sinter–crystallisation behaviour of G1 glass constituted the basis for preliminary experiments on macrocellular glass ceramics, by the sintering of glass powders around sacrificial polymeric templates. The high residual porosity and limited density increases accompanying increases in sintering temperature and time provide evidence that the crystallisation, mainly related to formation of fluorapatite or oxyfluorapatite (see Fig. 2), ‘freezes’ the viscous flow of the glass. This action impeded the viscous collapse of the cellular structure modelled by the PE templates, as illustrated by Fig. 4b. The homogeneity of the structure, higher than that of analoguous glass/PE mixtures, obtained without binders, as reported in the literature,30 was probably favoured also by the introduction of the silicone resin, joining the glass powders up to high temperatures.
Programming the multistep thermal treatment to end with a 30 min holding stage at 1070°C was conceived in order to favour the crystallisation, mainly of fluorapatite or oxyfluorapatite, more than the densification. The structural effect of this choice is shown in Fig. 4c: besides openings between macropores, also micropores, due to incomplete viscous sintering, are evident on the cell walls. Glass ceramic foams from G1 may be consequently considered as cellular materials with hierarchical porosity;31 such a particularly shaped open porosity could be advantageous in the view of applications as filters or catalyst supports, or as biomaterials, favouring the absorption interactions with bodily fluids.
The crushing strength of cellular glass ceramics, of ∼8 MPa, compares favourably with compressive strengths of other foam glass ceramics produced from waste21,32 and confirms observations concerning the strength of samples from simple sinter–crystallisation. As suggested by Gibson and Ashby,19 the crushing strength of open celled foams is proportional to the relative density and to the bending strength of the solid phase as shown in equation (1)
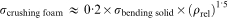
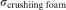 is the crushing strength of foam,
is the crushing strength of foam,
 is the bending strength of solid material and ρrel is the relative density. With a total porosity of ∼70% and, consequently, a relative density ρrel of ∼0·3, the bending strength of the solid phase associated with the observed crushing strength is ∼240 MPa, a value even greater than that reported for pore free apatite–wollastonite glass ceramics.6 It must be noted, finally, that 8 MPa is also quite close to the highest values reported for trabecular bone, of ∼10 MPa;33 the authors believe that this threshold value is reasonably achievable by optimising the proportions between glass and PE or by adjusting the sintering temperature and time.
is the bending strength of solid material and ρrel is the relative density. With a total porosity of ∼70% and, consequently, a relative density ρrel of ∼0·3, the bending strength of the solid phase associated with the observed crushing strength is ∼240 MPa, a value even greater than that reported for pore free apatite–wollastonite glass ceramics.6 It must be noted, finally, that 8 MPa is also quite close to the highest values reported for trabecular bone, of ∼10 MPa;33 the authors believe that this threshold value is reasonably achievable by optimising the proportions between glass and PE or by adjusting the sintering temperature and time.
The properties of larger samples obtained from G2 sintered at 800°C for 60 min (120 min did not lead to any significant change) are reported in Table 2. The residual porosity is lower than 3%, and this is confirmed by the micrograph in Fig. 5a (polished surface) and by the fact that the material is white with a certain degree of translucency, visible in Fig. 6 (the fragment is ∼3 mm thick). The formation of a silicate crystalline phase is the likely cause of the observed remarkable densification. As noted previously, the formation of combeite caused the glassy phase in G2 to be rich in P2O5. Phosphate glasses are known to generally exhibit lower viscosities than silicate glasses at a given temperature, albeit with some exceptions.34 The low sintering temperatures of 700–800°C were evidently high enough for a significant flow of the residual glassy phase. This behaviour is substantially the opposite of the case of G1, in which the first crystalline phases which developed were phosphate based, thus leaving a silicate residual glass with a higher viscosity that limited viscous flow.

Microstructural details of glass ceramics from G2 glass
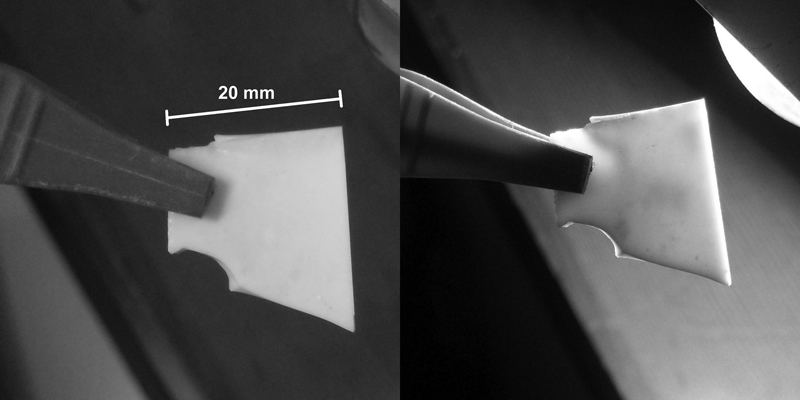
Qualitative evidence of translucency of glass ceramic from G2 glass
Figure 5b, showing the polished sample surface of G2 after HF etching, confirms the remarkable crystallinity of the obtained combeite based glass ceramic. The low porosity and high crystallinity are the reasonable causes of the interesting values of elastic modulus, bending strength and Vickers microhardness. The Young's modulus, being ∼90 GPa, is of the same magnitude as the values reported for other dense glass ceramics.6 The bending strength and the microhardness, exceeding 100 MPa and ∼6 GPa respectively, are also typical of dense glass ceramics employed for building applications or for dental restoration.6 In particular, the microhardness is higher than that reported by Abo‐Mosallam et al.18 for glass ceramics based only on combeite.
Owing to the observed mechanical properties the combeite glass ceramics could find profitable applications in the field of construction materials, in the form of tiles, or even in load bearing bone implants.
Conclusions
The authors conclude that the research described in this work can be summarised as follows.
Meat and bone meal ash, mixed with SLS glass cullet and secondary additives, can be readily vitrified to form silicophosphate glasses that in turn can be easily converted into apatite–wollastonite and combeite glass ceramics.
Biocompatible glass ceramics may be produced by rapid sinter–crystallisation treatments of fine glass powders obtained from vitrification of MBM ash with SLS glass scrap.
The glass leading to apatite–wollastonite glass ceramics features a remarkable crystallisation before the completion of densification by viscous flow sintering. This causes the sintered apatite–wollastonite glass ceramics to be porous, and it is also advantageous for limiting viscous collapse of macrocellular glass ceramics manufactured by sinter–crystallisation of glass powders mixed with PE templates.
For apatite–wollastonite glass ceramics, the rapid crystallisation of apatite leaves a residual silicate glassy phase with a relatively high viscosity; for combeite glass ceramics, the crystallisation of a silicate phase leaves a phosphate rich residual glassy phase capable of significant viscous flow even at low sinter–crystallisation temperatures, with the effect of greatly reducing the residual porosity and determining strong glass ceramic products.
The apatite–wollastonite and combeite glass ceramics developed here all exhibit mechanical properties that are highly desirable for a range of potential applications, including structural ceramics and biocompatible materials.
The combeite glass ceramics developed here require low sintering temperatures and short sintering times, enhancing the economic viability of processing such materials for use as, for example, bulk structural materials.
Footnotes
Acknowledgements
EB acknowledges Professor G. Scarinci for a very fruitful and stimulating discussion and Dr Paola Palmero (Politecnico di Torino, Turin, Italy) for supplying PE spheres. PAB acknowledges with thanks the assistance and warm hospitality of Dr E. Bernardo, Professor P. Colombo and their colleagues at Università di Padova. He also thanks Professor I. M. Reaney and EPSRC for supporting this research collaboration through grant no. EP/F012403/1.