
Abstract
In this study, Ca–Al–NO3 layered double hydroxide (LDH) nanoparticles of varying sizes were synthesised by a process involving co-precipitation under hydrothermal condition. This method produces stable homogeneous LDH suspensions under variable hydrothermal treatment conditions with particle size in the range of 7·5–2·5 μm. Layered double hydroxides were characterised by X-ray diffraction, Fourier transform infrared spectroscopy, thermal gravimetric/differential thermal analysis, scanning electron microscopy and transmission electron microscopy (TEM) and nanosizer analyses. By increasing the hydrothermal treatment time, the crystallinity and the particle size of obtained LDH increased. Scanning electron microscopy and TEM observations showed uniform hexagonal flake-like particles with high aspect ratio. Finally, Ca–Al–NO3 LDH did not show any acute cytotoxic effect up to 100 μg mL−1 as measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Introduction
Nanostructured materials have aroused considerable attention due to their unique physiochemical properties and their potential applications in a wide range. Among many nanostructured materials, two-dimensional layered materials offer a new area for developing hybrids with desired applications. Layered double hydroxides (LDHs), also known as hydrotalcite-like compounds, consist of hydroxide layers and anions. These class of anionic clay materials, represented by the generic formula [M(II)1−xM(III)x(OH)2]
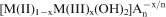 .YH2O, where M(II) and M(III) are di- and trivalent cations respectively.1 Structurally, positive brucite-like sheets are counterbalanced by the exchangeable interlayer anions, An−. The stability of structures including interlayer anions and water molecules is maintained by hydrogen bonds with the hydroxyl groups of octahedral sheets. Therefore, different compositions of LDH can be synthesised by a combination of M(II) (e.g. Mg, Ca, Ni, Zn, Co, Fe2+) and M(III) (e.g. Al, Cr, Fe3+) with different intercalated anions (e.g.
.YH2O, where M(II) and M(III) are di- and trivalent cations respectively.1 Structurally, positive brucite-like sheets are counterbalanced by the exchangeable interlayer anions, An−. The stability of structures including interlayer anions and water molecules is maintained by hydrogen bonds with the hydroxyl groups of octahedral sheets. Therefore, different compositions of LDH can be synthesised by a combination of M(II) (e.g. Mg, Ca, Ni, Zn, Co, Fe2+) and M(III) (e.g. Al, Cr, Fe3+) with different intercalated anions (e.g.
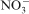 , Cl−,
, Cl−,
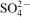 ,
,
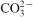 ).2,
3
).2,
3
Owing to the versatile properties of LDH, i.e. good biocompatibility, swelling, pH dependent solubility and anion exchange capability, this group of materials is potentially suitable for controlled release of carried drugs, cellular delivery and biomolecule protector systems.4–6
One of the most commonly used methods for synthesising LDH is co-precipitation at variable or constant pH, followed by aging at predefined time and temperature.7 These synthesised LDHs tend to be aggregated in aqueous suspension with a secondary particle size of 1–10 μm, not suitable for cell related applications.8 Since most biological interactions take place in nanoscale, it is necessary to obtain homogeneous, stable, nanocrystalline LDH. Therefore, LDH particle size control is an important key when LDH materials are used for cellular drug delivery or thin film preparation. There are some reports on synthesising homogeneous LDH suspension with controllable particle size.9, 10 Choy et al.11 evaluated hydrothermal treatment for tailoring the Mg–Al–CO3 LDH particle size, while some aggregation of particles has been reported. Some researchers employed the urea hydrolysis method to produce Mg–Al–CO3 LDHs in sizes 1–10 μm, which is not appropriate for cell related applications.12, 13
Since hydrothermal treatments could generally affect the particle size and crystallinity of the obtained LDHs, the LDH configuration of particles can be changed to big flakes. Kovanda et al.14 investigated the different factors that affect the condition of synthesis (e.g. aging time, temperature and concentrations) in order to control the particle size of LDHs.
In this work, hydrothermal treatment has been employed for preparing stable and homogeneous suspension with controllable particle size. We also investigate the in vitro cytotoxicity of Ca–Al–NO3 LDH on L929 mouse fibroblast cell line in order to show that LDHs can be potentially used in biosystems.
Experimental
Materials
Calcium nitrate tetrahydrate [Ca(NO3)2.4H2O] (Merck 13477-34-4), aluminium nitrate nonahydrate [Al(NO3)3.9H2O] (Merck 7784-27-2) and sodium hydroxide (NaOH) (Merck 1310-73-2) were used as starting materials for LDH synthesis. All chemicals were analytical grade and used with no further purification. Decarbonated water was obtained by boiling deionised water and was used in all of the processes.
Layered double hydroxides synthesis
Ca–Al–NO3 LDH powder has been synthesised by conventional co-precipitation technique as described previously.7 The synthesis was carried out at room temperature by dropwise addition, under constant stirring, of 50 mL solution A containing NaOH (6 mmol) to 100 mL solution B containing salts solutions of Ca(NO3)2.4H2O (2 mmol) and Al(NO3)3.9H2O (1 mmol). During the reaction, the pH of the slurry was kept at 10. It is worth mentioning that to avoid contamination of atmospheric CO2, all processes were performed under N2 atmosphere. Subsequently, the slurry was aged for 12 h at room temperature and simultaneously ultrasonicated. Eventually, the resulting precipitate was collected via centrifugation (4500 rev min−1 for 5 min) and washed three times with decarbonated and deionised water.
After the final centrifugation step, a known amount of pellet was resuspended in 50 mL deionised water and transferred to a Teflon autoclave. The autoclave was then placed in the oven, followed by hydrothermal treatment at a temperature of 120°C for 0, 4, 24 and 44 h (samples S0, S1, S2 and S3 respectively). After air cooling, a known amount of stable homogeneous suspensions was kept for further analysis, while the remaining suspension was freeze dried to obtain white powder.
Layered double hydroxides characterisation
X-ray diffraction
The structural characterisation and phase identification of the synthesised LDH were performed with a D4 Bruker X-ray diffractometer system with a monochromatic Cu Kα radiation (λ = 1·5406 Å) over the 2θ range of 5–50° at a scan rate of 2° min−1 in Guiner geometry. The operation voltage and current were 40 kV and 30 mA respectively.
Fourier transform infrared spectroscopy
Fourier transform infrared spectrum was obtained on a Bruker IFS 48 spectrometer. The potassium bromide disc technique was used for the analysis using 2 mg of LDH powder compacted with 200 mg of potassium bromide under a hydraulic pressure. The spectrum was recorded in the 4000–400 cm−1 region with 2 cm−1 resolution averaging 100 scans.
Particle size analysis
The average particle size (z average size) and size distribution were recorded by Nanosizer Nano ZS, Malvern Instruments. The same instrument was used for zeta potential measurement.
Scanning electron microscopy
The morphological observation was conducted by a XL30 Philips scanning electron microscope (SEM). The sample preparation for SEM analysis was performed by sprinkling the LDH powder onto one side of a double adhesive tape, which was stuck to an aluminium sample holder. The samples were coated with a thin layer of gold by sputtering (EMITECH K450X, UK) to a thickness of 20–30 nm and examined with an acceleration voltage of 20 kV.
Thermal analysis
The thermal behaviour of the LDH was studied using thermogravimetric analysis/differential thermal analysis (simultaneously thermal analysis; Polymer Laboratories PL-STA 1640) from room temperature up to 1200°C at a heating rate of 10°C min−1. It is important to point out that an α alumina crucible was used as reference in this analysis.
Transmission electron microscopy
The transmission electron microscopy (TEM) images were obtained on a Philips EM208 at an acceleration voltage of 200 kV. For sample preparation, the freshly prepared LDH nanoparticles were dispersed in alcohol with ultrasonication for 30 min, and then, a droplet was dropped on a copper grid coated with amorphous carbon film.
Biocompatibility evaluation
L929 mouse fibroblast cell line was used in this study. Cells were cultured in
Results and discussion
X-ray diffraction analysis
The X-ray diffraction (XRD) patterns for the samples S0, S1, S2 and S3 are shown in Fig. 1. All of the samples exhibit the Bragg reflections of basal planes. The series of (00l) peaks are symmetric at low 2θ angles but broad and asymmetric at high 2θ angles.10 Lattice parameters of samples are displayed in Table 1. The values of basal spacing were calculated by Bragg's equation from (00l) peaks using the 1/2(d002+2d004) formula. The parameter a is related to the cation–cation distance within the brucite-like layer a = 2(d110), where d110 corresponds to peak (110). The value of a depends on the nature of the cations.15 Parameter c is related to the thickness of the brucite-like layer and the interlayer distance according to hexagonal polymorphic form of Ca2Al LDH; the c parameter corresponds to two times the interlayer spacing, c = d002+2d004. The value of the basal spacing (0·86 nm) corresponds to the sum of the thickness of the hydroxyl layer ([Ca2Al(OH)6], 0·48 nm) and of the interlayer anions (nitrates), which is ∼0·38 nm. This is in agreement with the previous reported value for the basal spacing including the planar orientation of nitrates and water molecules within the interlayer space of LDH.16 The hydrothermal treatment results in increasing intensity of diffraction for the S1, S2 and S3 samples. All the samples demonstrated characteristic diffraction peaks of an LDH, and a relative sharpness of peaks can be seen in sample S3 with 44 h hydrothermal treatment time.
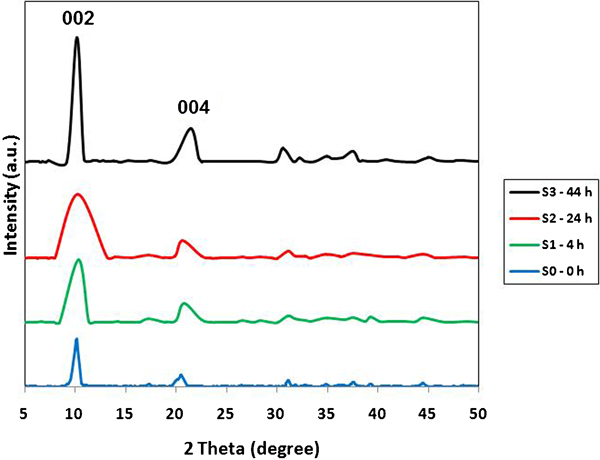
X-ray diffraction patterns for LDHs with different hydrothermal treatment times
Lattice parameters of LDH samples
Fourier transform infrared spectroscopy analysis
The Fourier transform infrared spectra of all the prepared LDHs are similar together. As shown in Fig. 2, the band ∼3450 cm−1 related to the O–H vibration mode of the hydroxyl group and water molecules. The peak at 1384 cm−1 assigned to the presence of the
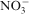 group. A characteristic frequency for Ca–Al–NO3 LDH is the bending vibrations of water molecules at 1600 cm−1. In the low frequency region of <1000 cm−1, the band of the spectra corresponded to the M–O vibration bonds. In addition, owing to the broadness of the nitrate band, it may also correspond to the overlapping of carbonateions with nitrate ion, which usually occurs at 1360–1370 cm−1.16 Moreover, the presence of the carbonate group in the infrared spectra may arise from contamination of the system by absorption of carbon dioxide from air during the synthesising process.
group. A characteristic frequency for Ca–Al–NO3 LDH is the bending vibrations of water molecules at 1600 cm−1. In the low frequency region of <1000 cm−1, the band of the spectra corresponded to the M–O vibration bonds. In addition, owing to the broadness of the nitrate band, it may also correspond to the overlapping of carbonateions with nitrate ion, which usually occurs at 1360–1370 cm−1.16 Moreover, the presence of the carbonate group in the infrared spectra may arise from contamination of the system by absorption of carbon dioxide from air during the synthesising process.
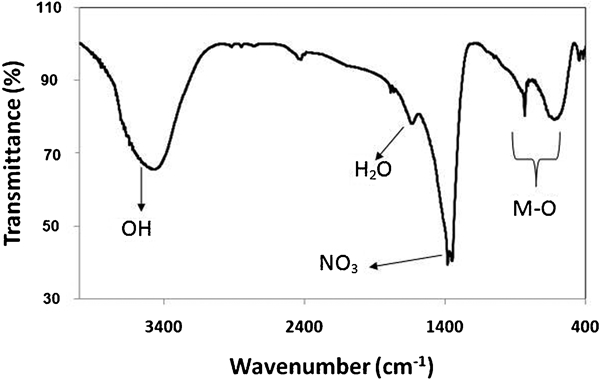
Fourier transform infrared spectroscopy spectrum of typical Ca–Al–NO3 LDH
Particle size distribution
The particle size distribution and the peak value are highly dependent on the hydrothermal treatment technique. Figure 3 shows that in the untreated S0 sample suspension, the particle size distribution is broadened from 100 to 2669 nm, which corresponds to aggregation of LDH crystallites. In contrast, in samples that experienced 24 and 44 h hydrothermal treatment, the LDH particles become uniform, with a narrow particle size distribution. It can be seen that the 4 h treatment in sample S1 results in smaller particle size (7·5–78 nm) comparing with the S2 and S3 samples; it is due to the full dispersion of aggregates into crystallites that happened in the early stage of treatment. Increasing hydrothermal treatment time leads to growth and reaggregation of particles; therefore, the 44 h treated LDH suspension contains much bigger particles with size distribution of 105–2305 nm. This phenomenon indicates the continuous growth of LDH particles with time and reaggregation of particles.
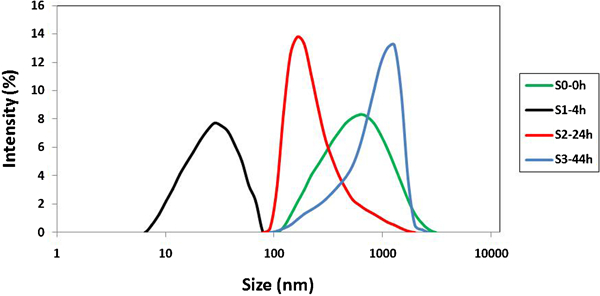
Particle size distribution of Ca–Al–NO3 LDHs with 0, 4, 24 and 44 h hydrothermal treatment
Scanning electron microscopy and TEM observations
To observe the influence of hydrothermal treatment on morphology of LDH, SEM technique was employed. Figure 4a and b shows the morphology of the S0 sample. As can be seen in this figure, in the absence of hydrothermal treatment, particles are highly agglomerated with some amorphous conglomeration (see black arrows). When samples are heated, the amorphous regions disappear and the size of the platelets seems to enlarge. As can be seen in Fig. 4c and d, the particle dimension of the 24 h hydrothermal treated sample is larger than that of the untreated sample. By increasing hydrothermal treatment time, for sample S3 (Fig. 5), the crystal particle size grows bigger, the hexagonal flake-like shape becomes more regular and aspect ratio increases. This phenomenon can be well observed for sample S3 in comparison with S0 and S2 samples. In TEM images of Fig. 6a, the LDH crystallites (sample S1) are very well separated and well shaped in hexagonal form with a lateral size of ∼10 nm.

Images (SEM) of samples with magnifications of a ×5000 and b ×15 000 for S0, and c ×5000 and d ×15 000 for S2

Images (SEM) of 44 h hydrothermal treated sample (S3), with different magnifications of a ×5000, b ×10 000, c ×15 000 and d ×20 000
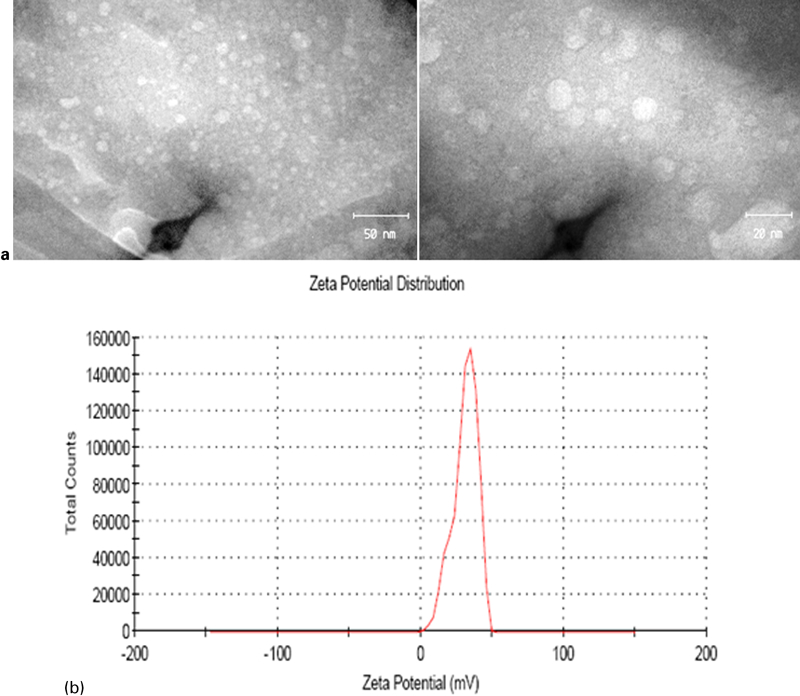
a images (TEM) and b zeta potential curve of Ca–Al–NO3 LDH with 4 h hydrothermal treatment (S1 sample)
As shown in Fig. 6b, the zeta potential of the S1 sample was measured as 30·9 mV. This high charge on the surface of LDH makes it suitable to adhere to cell membrane and cellular uptake.
Thermal analysis
The differential thermal analysis and thermogravimetric analysis (simultaneous thermal analysis) curves of the Ca–Al–NO3 LDH are illustrated in Fig. 7. In general, three main endothermic effects were found during the compound decomposition and weight losses. The first process was up to the temperature of 150°C and is due to the removal of physically adsorbed water molecules on the surface and interlayer space. The second process continued up to 330°C and is associated with dehydroxylation of the structure and reduction in nitrates to nitrites. The third endothermic peak occurring up to 530°C is related to a further removal of hydroxyls and decomposition of nitrites. The thermogravimetric curve demonstrates complete decomposition of the sample at 600°C, and the total weight loss is 48%. The thermogravimetric curve of the compound also shows three main steps, as mentioned above. From room temperature to 150°C, the compound loses 9·9% of its weight. The second weight loss is up to 330°C and corresponds to 24% of total weight. The weight loss between 330 and 600°C is mostly consistent with thermal decomposition of nitrate anions. As reported in the literature, despite the fact that most water molecules disappear from the sample, the basal spacing is not changed.16–18 In the first stage, dehydration occurs as follows
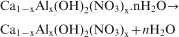
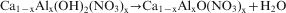
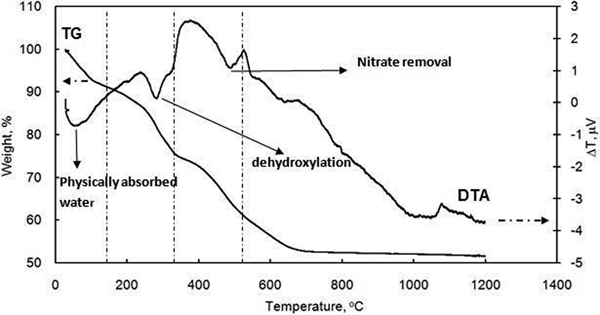
Thermal analysis curves of typical Ca–Al–NO3 LDH
Biocompatibility evaluation
For the viability study, cells were exposed to various concentrations of LDH particles, which ranged from 0 to 500 μg mL−1. It was found that over a 4 day period, the viability of the cells remained ∼80% within concentration up to 500 μg mL−1. On the other hand, after 6 days of incubation, a remarkable loss of viability was observed at concentrations of 100, 200 and 500 μg mL−1. As shown in Fig. 8, 500 μg mL−1 of LDH significantly reduced cell viability after 4 and 6 days.
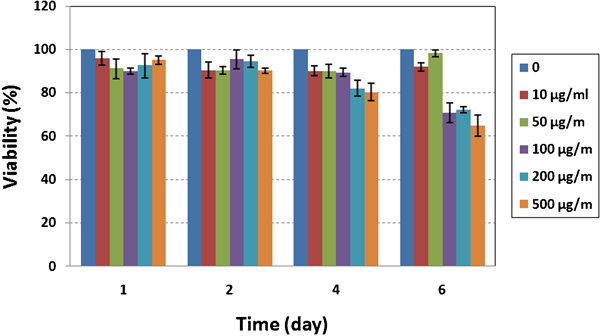
Cytotoxicity of LDH nanoparticles on L929 mouse fibroblast cell line (tests were performed in triplicate, and data were reported as average; optical density of samples was normalised to growth control)
The viability of cells treated with 10 and 50 μg mL−1 of LDH did not significantly change from 1 to 6 days but decreased at higher concentration after 6 days. It is interesting to note that the viability of LDH increased at a concentration of 50 μg mL−1 from 1 to 6 days; the reason for which is not clear. Also during cell treatments, no cell detachment or rounding was observed.
Reaction mechanism
Generally, during the hydrothermal treatment, a series of processes including disaggregation of particles, particle regrowth and reaggregation take place. Dissociation, deposition and diffusion (disaggregation) are the main events within the first 2–4 h of hydrothermal treatment.19 By dissociation, the amorphous phase of LDH is dissolved. Thus, the glue effect of the amorphous phase that results in forming aggregates reduces.9 By diffusion, metal cations (Ca and Al) are dispersed within layers in order to reduce the lattice defects and enhance the crystallinity of LDH. On the other hand, by increasing the temperature in the hydrothermal treatment, the Brownian motion of particles increases, so that LDH crystallites could escape from the aggregation. It could be believed that after the escape of the LDH crystallites, they will tend to repel each other due to the electrostatic repulsion of positively charged particles (positive zeta potential). Therefore, a stable and homogeneous suspension can be obtained after 4 h of treatment. Following disaggregation, the growth phase of the LDH crystallite begins.
It is to be noted that LDH crystallite growth occurs in parallel with disaggregation in the early stages of hydrothermal treatment (2–4 h). After complete disaggregation, dissolving small crystallites and depositing the cation (Ca and Al) species onto the bigger crystallites lead to better crystallisation.
Consequently, continuous growth of particles results in reaggregation because these big particles could overcome the electrostatic repulsion.
Furthermore, hydrothermal treatment time highly affects on particle size distribution and dispersion state. Meanwhile, increasing time of treatment from 4 to 44 h leads to much bigger particles. Suspension of freshly prepared Ca–Al LDH (S0) consists of broad particle size distribution ranging from 122 to 2·7 μm.
Fast co-precipitation makes small LDH crystallites quickly aggregate due to the presence of the amorphous LDH phase and sharing of surface anions within the overlapping area between two sheet-like crystallites and sharing of edges of different crystallites.20 In fact, fast co-precipitation quickly leads to a number of uniform nuclei (crystallites) to interconnect by sharing edges and surfaces of the sheet-like crystallites to form aggregates. Compared with the freshly prepared LDH suspension, the 4 h treated LDH suspension contains much smaller particles that are 7·5–70 nm in size. This indicates that particles are dispersed into individual crystallites, but they are not reaggregated. It could be believed that when treatment time increases, high pressure restricts the growth direction (c axis direction) for new crystallites and crystallites nucleate and grow along the a and b axes because the formation of M–OH valence bond thermodynamically is more preferred than formation of layer along the c axis. In this case, metal oxides are dissolved, and the nitrates intercalate into the interlayer space of LDHs; subsequently, recrystallisation occurs. Indeed, in the early time of hydrothermal treatment, disaggregation happens, after which the growth of the LDH individual crystallites takes place; whereas for the short treatment time (2–4 h), reaggregation does not take place. For example, the treatment at 100°C for 4 h disperses the aggregates, while the treatment at same temperature for 24 h results in bigger LDH sheets with micrometer scaled particles. As the treatment time increases further, the LDH particle size distribution is shifted to the larger particle size, indicating the continuous growth of LDH particles with time.21–23
Conclusion
In summary, Ca–Al–NO3 LDH was successfully synthesised by the co-precipitation method followed by hydrothermal treatment. This technique can be used to control particle size in the range of 10–2·5 μm in the lateral dimension and make stable LDH suspensions. Moreover, the LDHs with nanoscaled crystallites were synthesised at 120°C with hydrothermal time of 4 h. Finally, a stable suspension of LDH with nanosized particles can be useful as a reservoir and carrier for drugs, gene and biomolecules.
Footnotes
Acknowledgements
This work was financially supported by the Iran National Science Foundation.