
Abstract
The effect of high energy dry milling on the structural and crystalline state of sintered biphase calcium phosphates was studied. After various periods of grinding, the initial biphase calcium phosphate material alters its crystalline structure and phase composition. The phase transformations achieved during milling were recorded by powder X-ray diffraction, scanning electron microscopy, infrared spectroscopy and chemical analysis. X-ray diffraction analysis of samples milled for 20 h showed that the initial composition of the biphase ceramics changed and part of β-tertiary calcium phosphate was nanocrystalline and partially in amorphous state. Hydroxylapatite fully transformed into nanocrystalline phase. Cytotoxicity tests of samples milled for 20 h clearly present cellular viability with pronounced biological activity.
Keywords
Introduction
Recently, there is a constantly growing interest in sintered biphase calcium phosphates (BCPs) hydroxylapatite (HAP) and β-tertiary calcium phosphate (β-TCP) used as scaffold materials, with chemical composition close to the mineral part of the bone (with molar ratio of Ca/P near 1·67) and solubility favourable for the biomineralisation around the bone implant materials.1 It is known that BCP materials are more effective in bone repair and regeneration than monophase HAP or β-TCP ones, and have a controllable degradation rate to a certain degree.2–7 Composites of β-TCP and HAP biphase phosphates combine the excellent bioactivity of the two phases: the good osteoconductivity of HAP and the high properties of resorbability (bioresorbability) of β-TCP.8, 9 Thus, these different properties of the two phases give impetus for study of such biphase type of Ca phosphates. Understanding the synthetic characteristics and structural chemistry of calcium phosphate materials is a key into their improved medical application, but their chemistry is complex, offering diverse structure with intrinsic compositional variation.10 In this point of view, there are a lot of possibilities to approve the quality of advanced biomaterials. In order to apply appropriate BCPs to meet specific biological need, it is crucial to control BCPs with various ratios of HAP/TCP. Furthermore, recently, nanobioceramics have attracted attention for their effective bioactive properties.11–16 Many studies have shown that bone forming cells for specific application are interacting with nanoscale surfaces of biomaterials, which is critical to keep the body from rejecting artificial parts17, 18 and promote the adhesion, proliferation and differentiation of osteoblasts.19, 20 Nevertheless, the relationship between the characteristics of crystals and their biological activity is still unknown. Despite difficulty in accurately characterising the composition, the structural details and the properties of calcium phosphate nanocrystals, the investigations in this direction make intense progress. It is essential to develop the use of apatite nanocrystals in biomaterials and to reach a better understanding of the role and behaviour of the bone mineral.21 Some physicochemical parameters such as surface topography, particle size and tridimensional structure of biomaterials are factors in bone regeneration and repair processes. Similar situation exists in the fields of nanocrystalline apatite considered to play a major role in the biologically active materials,21, 22 but they remain rather poorly defined and characterised. The poor knowledge and characterisation of calcium phosphate nanocrystals may result in inconsistencies and disagreement in experimental results.
Mechanical milling of solid state material is a method that takes advantage of the perturbation of surface bonded species by pressure to enhance their thermodynamic and kinetic reactivity. In addition, it is one of the promising techniques to receive nanosized material. Different morphologies, stoichiometries and crystallinities can be obtained by mechanical treatment processes depending upon the technique and the materials used.23–28 Dispersion in grinding leads to radical changes in microstructure. Not only does grinding increase the surface area of particles, it also changes the state of the surface layers. In addition, defects accumulated in the volume and different reaction centres are formed at the rupture zones.29
It is well known that one of the most interesting features of mechanical milling is the ability to produce nanosized powders. Deformation strain during milling depends generally on the mechanical treatment conditions and the nature of treated materials. As a result of increasing the milling time, the crystallite size decreases until a steady state particles size is approached.
To the best of our knowledge, at present, there is distinct lack of studies in the high energy dry grinding of BCPs and the data considering that the results of structural changing and phase transformations of this material are missing. From such point of view, it must be mentioned that single phase β-TCP subjected to high energy grinding for 20 h gets fully amorphous30 and single phase HAP at the same conditions is transformed into nanocrystals. Up to now, there are still no data about phase transformations of BCPs during milling especially for transformation in nanostate of both HAP and β-TCP synthesised as new bone implant materials with complex 3B activity (bioactivity, biocompatibility and bioresorbability). In this context, it is of growing interest to study such processes in order to obtain biomaterials with new complex properties.
The present work is based on a promising approach by use of dry milling process to prepare BCP nanomaterials with different morphologies and functionalities.
The aims of the present study are to make attempt to evaluate the effects of high energy dry grinding of BCPs for different lag times until obtaining nanometric crystal size materials. For this purpose, BCP sintered samples are synthesised and characterised. Subsequently, they are subjected to high energy dry milling. The obtained materials are then studied by chemical, morphological and structural analyses.
Experimental
Materials
The starting material used in this study was poor crystalline apatite or non-stoichiometric apatite, synthesised by double decomposition of Ca(NO3)2.4H2O (1M in deionised water) and (NH4)2HPO4 (0·6M in deionised water) (Merck) to resave solutions with molar ratio Ca/P = 1·67. The phosphate solution was dropwise poured with a rate of 4 mL min−1 into the solution of Ca (NO3)2 at constant stirring and continuing keeping pH about 10·5–11 (adjusted by addition of NH4OH) at room temperature. The mixture was maintained in solution for 24 h at room temperature for aging, ensuring that the reagents were fully reacted and precipitated. The chemical reactions may be presented according to
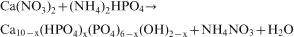
Depending on the degree of Ca deficiency x, the molar composition of the resulting biphase powder mixture of the calcinations >750°C can vary according to the following equation31,
32
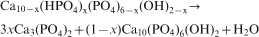
Subsequently, the sintered sample A1 (at 1100°C) was dry milled in agate planetary ball mill (‘pulverisette’ 6 Fritsch) at 600 rev min−1, with agate bolls (10 mm diameter) in standard mass of sample ∼30 g. The milling process was carried out for periods of times of 5, 10 and 20 h to obtain nanocrystalline sizes of the material. The mechanochemical treatment (high energy milling) increases the material specific surface area and changes the material structure, generating dislocation and point defects, which favour microarea reactivity. A small quantity of powder was taken from the mill chamber during different periods of time to be analytically investigated. The obtained milled samples are denoted as A1–B5, A1–B10 and A1–B20. This nomenclature is given in Table 1.
Nomenclature of studied samples

Methods
The following methods were employed to study the phase transformation and physicochemical characteristics of all studied samples.
The crystalline phases in all samples were determined by powder X-ray diffraction (XRD) with a Bruker D8 diffractometer, operating at 40 kV and 40 mA with Cu Kα radiation. This method was used to identify the crystalline phases present in the milling materials. Scans were performed over the range of 2θ 10–80° (step size, 0·01; counting time, 1 s).
The infrared (IR) spectra were collected by a Tensor 37 spectrometer (Bruker) and collected with a 4 cm−1 resolution after averaging 72 scans on standard KBr pellets in the spectral region of 400–4000 cm−1 at room temperature. The powders were mixed with IR quality KBr at a mass ratio of ∼1∶400, and the mixture was pressed into pellet. The adsorption spectra were acquired over the range of 400–4000 cm−1 with a resolution of 2 cm−1. Each spectrum was scanned 32 times to increase the signal/noise ratio. The estimated standard deviation of the wavelength was 4 cm−1.
Microstructure and morphological characterisation of the milled materials was studied on a JEOL JSM-5510 at a beam current of 20 mA and an accelerating voltage of 20 kV.
For chemical analysis, calcium concentration was determined by complexometry with EDTA and phosphorous concentration was determined by spectrophotometry as a phospho-vanado–molybdenum complex using the spectrophotometer NOVA 60 (Japan). The Ca/P atomic ratio of the prepared material was calculated from the obtained results. The measurements were carried out using 50 mg powder dissolved in 20 mL water solution of HNO3.
The cytotoxicity test of the material was performed in Dulbecco's modified Eagle's medium (DMEM) according to the extraction cytotoxicity test: ISO 10993-5:2009(E) to determine the cellular viability. Test samples for cytotoxicity of ground material denoted as tests-CP were prepared from 100 mg of the milled material mixed with 0·33 mL double distilled water placed on glass slide 5 cm2 with 10 mL DMEM in Petri dish with d = 10 cm and incubated for 24 h in accordance with ISO 10993-12. The non-tumour permanent cell line Lep-3 from a 3 month embryo was used as an experimental model. The cells were routinely grown as monolayer in DMEM supplemented with 5–10% foetal bovine serum, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cells cultured in DMEM media were used for control. The experiments were performed during the exponential phase of cell growth, approximately at 80% confluent state.
Lep-3 cells were seeded in 96-well flat bottomed microplates at concentration of 104 cells per well. After the cells were grown for 24 h to a subconfluent state, 60–70% of the cell monolayer was covered with 100% t-CP (test medium). Control cells were cultured in DMEM without t-CP. Cell viability was subsequently studied in t-CP media diluted with pure culture media in ratios of 3∶1, 1∶1 and 1∶3 (v/v) 75, 50 and 25% respectively for t-CP media. After 72 h of incubation, the effect on cell viability and proliferation was studied by the following:
thiazyl blue tetrazolium bromide (MTT) test, colorimetric assay of cell survival33
neutral red (NR) uptake cytotoxicity assay was based on the method of Borenfreud and Puerner34
crystal violet (CV) staining was based on the method of Saotome et al.35
Relative cell viability expressed as percentage of the untreated control (100%) was calculated for each concentration of t-CP. Statistical analysis of the results was performed by one way analysis of variance followed by Dunnett post hoc test.
Results and discussion
Phase evolution in sintered poor crystalline non-stoichiometric apatite
Structural information obtained by XRD during sintering processes is essential for the history of the material used in this study for following the subsequent effects of the high energy grinding processes on the sintered sample (A1).
The main phase obtained after sintering of the initial precipitate (Ao) at temperature of 800°C is apatite denoted as A, and in the biphase sample at 1100°C (denoted as A1), the phases are HAP and β-TCP. The powder XRD patterns of the samples milled for 5, 10 and 20 h and A1 are presented in Fig. 1.
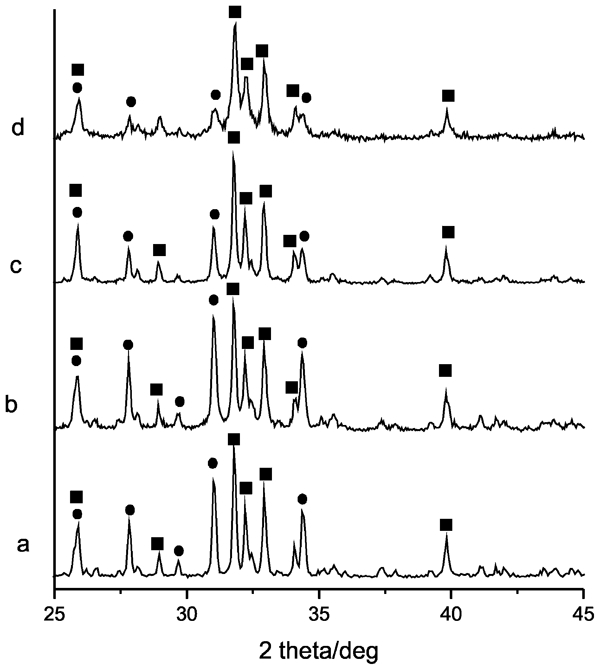
Powder XRD patterns of simple A1 and this sample milled for 5, 10 and 20 h (A1–B5, A1–B10 and A1–B20 respectively). squares indicate HAP and circles indicate beta-TCP
Having in mind the complexity of the processes involved in the precipitation synthesis, it is obvious that understanding the mechanism in the individual history of the samples used is an intriguing task.
For the interpretation of the kinetic results of calcinations in these experiments, temperatures of 800 and 1100°C were chosen. This model was chosen in the context of the data discussion in the literature for the formation of biphase β-TCP/HA materials. Upon calcinations of Ca deficient apatite, there are difficulties in accurate determination of the temperature at which phase transformations occur.36 The results obtained during heat treatment are difficult to interpret without kinetic study.37
The powder XRD patterns of the sample at 800°C are characterised by diffraction peaks identical to the patterns of HAP. This is due to the fact that the Ca deficient apatite has an HAP precursor in its structure36 but also is a result of a series of crystalline intermediate phase transformations involved in the structural and compositional changes.38–43 The phase transformation of Ca deficient apatite into BCPs (HAP and β-TCP) was demonstrated by XRD for samples sintered at 1100°C as well. The calcined sample was subsequently subjected to high energy dry milling.
The effects of kinetic dependences of the material phase transformation and chemical behaviour during formation of BCPs are not fully understood and deserve further investigations. Despite the recent advances in the knowledge of their structure and physicochemical behaviour, a number of important aspects still have to be considered, which will help to model the performance of this material.44
Powder XRD characteristics
It is well known that the most interesting features of the mechanical milling are to produce nanostructure powders. With prolonged milling, the material transformed to amorphous and nanocrystalline phases. As a result of increased milling time, the crystallite size of the material decreases continuously until some lower size limit when many defects in the crystal lattice are accumulated.45 Structural information obtained by XRD for samples milled for 5, 10 and 20 h is essential in the interpretation of the effects of high energy grinding taking place during milling at different periods of time. For all samples, the XRD diffraction peaks are identical to the characteristic ones of HA and β-TCP.
Table 2 summarises the results obtained by powder XRD analysis for sample A1 after milling for 5, 10 and 20 h. It is seen that the intensity ratio between the most intensive XRD peaks of HAP (211) and β-TCP (02·10) peaks increases from the samples sintered at 1100°C A1 in comparison with the milled ones for the various time periods used. The crystallite sizes gradually decreased upon milling. The observation also indicates that the crystalline amount of β-TCP phase decreases faster upon milling treatment, and subsequently, with operating time, a greater part of it gets amorphous. The effects of mechanical activation may be attributed not only to crystallite size decrease but also to mechanically induced transformation from crystallite to amorphous state.46–48
X-ray diffraction data about determined intensity of diffraction lines as area below the peak, intensity ratio of strongest lines of HAP and β-TCP, crystallites size, and full width at half maximum for (211) and (02·10) peaks for samples sintered at 1100°C and milled for various time periods
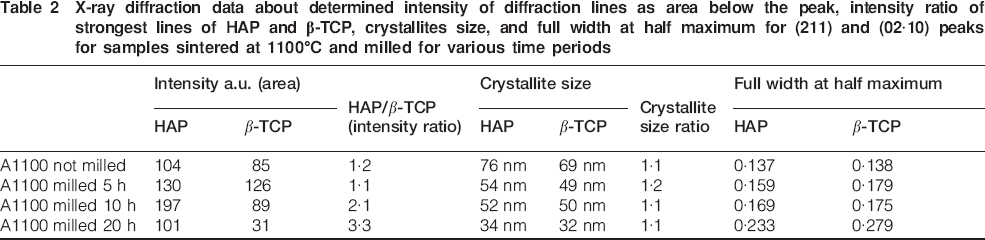
The increase in full width at half maximum of the peaks is negligible after 10 h of treatment and significant after extending time of milling 20 h, which is an indication of amorphisation of β-TCP (seen from the intensity ratio HAP/β-TCP, Table 2) The crystallite size of the initial and milled samples was determined using the WinFit software,49 and the obtained results show interesting tendencies. Both HAP and β-TCP decrease in crystallite size, uniformly confirmed by the equality in the crystallite size ratio being ∼1∶1. The refinement of the unit cell parameters of HAP and β-TCP in the studied samples was performed with the PDI package50 using non-linear least squares procedure for minimising the SUM [Q(exp)−Q(cal)]2/1/d2. The obtained values for the unit cell parameters for the two phases are given in Table 3.
Values for refined unit cell parameters for two phases for different milling times
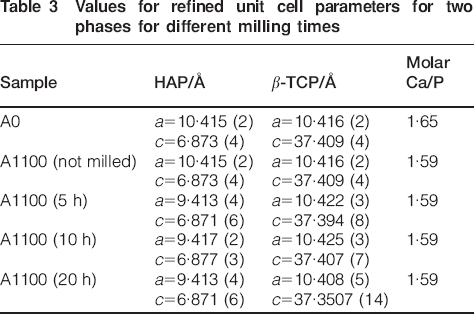
Analysis of the results presented in Table 3 shows that the unit cell parameters of both phases decrease. The observations agree with the interpretation of high energy milling as a progressive deterioration of original crystalline structure with production of extended dislocations, defects and some partial decomposition of the solids. As a result of this process, the defects and vacancy concentrations increase.
Nevertheless, for β-TCP milled at 20 h, there is observed apparent decrease in lattice parameters from the initial [a = 10·416 (2) Å and c = 37·409 (4) Å] to noticeable diminishing after milling a = 10·408 (5) Å and c = 37·3507 (14) Å. It is important to mention that after 20 h milling, a great part of β-TCP gets amorphous and the remaining is partially crystalline. The crystalline part has distinctly changed its lattice parameters. The structural changes probably involve changes/distortions in PO4 tetrahedrons, significant enough to result in changes in lattice parameters. Likewise, it is probable that these changes should be also assumed as a result of the specific structural changes of β-TCP related to cationic and anionic replacements. The cell parameters of the partially crystalline phase of β-TCP decrease with milling. The crystallite sizes for both phases HA and β-TCP get smaller as a function of milling time (Fig. 2).
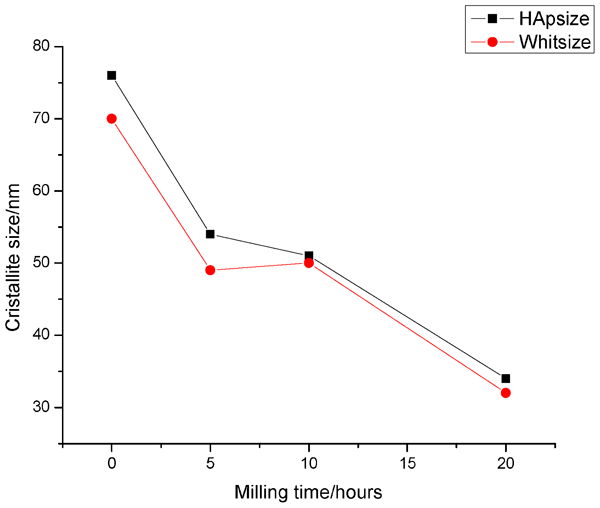
Calculated nanosized particles for both phases (HAP and β-TCP present in BCP sample) at beginning and as function of lag time of milling
It was found that the crystal sizes decreased considerably (calculated from powder XRD data). At the beginning, they have values 70 and 77 nm for the crystalline part of (TCP) and for (HAP) and ∼36 and 30 nm at the end of the experiment, after 20 h milling (Fig. 2).
The analyses of the concentrations of Ca and P in all samples under study are used to follow the changes of molar ratios Ca/P. These results are presented in Table 3. Analyses of these results show that the initial sample shares the molar ratio of the poorly crystalline apatite structure Ca/P = 1·65, which after sintering at 1100°C changed to 1·59 following the phase transformation into biphase material of HAP and β-TCP. After milling, there was no change in the calculated molar ratio, and for all samples, it is Ca/P = 1·59.
The results obtained in this study are of interest for syntheses of new advanced biomaterials because their functions such as solubility, dissolution rate and bioreactivity are generally controlled by their phase structure, crystallinity, crystal size and chemical composition. On the other hand, it was demonstrated that high energy milling of BCP is a useful approach for preparation of nanosize BCP material with unique characteristics of nanocrystals of HAP and β-TCP, partially presented in amorphous and also in crystalline forms.
Microstructure and morphological characterisation of sintered and milled Ca–P materials
The morphology of the high energy grinded samples was characterised by SEM technique. The presented micrographs on Fig. 3 show microparticles characteristic for BCP samples sintered at 1100°C (A1) and dry milling for 20 h. Foamy agglomerated particles and nanometric grains are observed with a complex nanoscale surface structure with mean sizes about 50–100 nm. The agglomerated particles are of several micrometres with randomly distributed nanosized voids and are associated with nanoparticles at their surfaces, with rough non-uniform forms and borders. All photographs show polymorph nanoparticles preferably with spherical shape and foam-like agglomeration.

The results from SEM confirmed the complex characteristics of the morphologies of phase transforming BCP as a consequence of high energy dry milling. A progressive loss of the original crystalline morphology was shown to occur. Resulting particles with irregular surfaces and rounded edges are seen in SEM. These observations agree with the mechanical treatment as a progressive deterioration of the original crystalline structure with production of extended defects and partial decomposition of the solids. These observations are in agreement with the results discussed above from our XRD measurements. It is of interest that parameters such as particle agglomerates, surface topology, particle geometry and nanometric size of the crystals should have a potential role on various biological performances, although the exact function of these morphological parameters on bone repair has not been unambiguously discussed.51, 52
Infrared spectroscopic analysis
To study the changes of
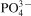 group and others such as OH−,
group and others such as OH−,
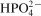 and
and
 in samples of BCPs during dry milling for 5, 10 and 20 h, FTIR spectroscopy was used. Table 4 summarises the results from the IR study.
in samples of BCPs during dry milling for 5, 10 and 20 h, FTIR spectroscopy was used. Table 4 summarises the results from the IR study.
FTIR absorption bands (cm−1) of control sample and samples dry milled for 5, 10 and 20 h
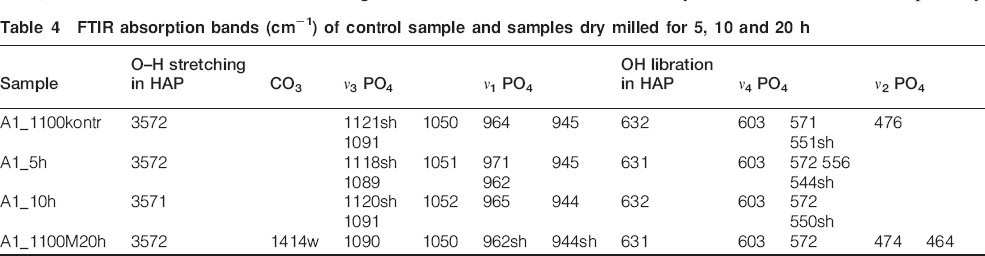
In well crystalline apatite, there are clearly resolved ν1 and ν3 peaks. The effect of the high energy dry milling treatment of samples of HAP/β-TCP on the absorption spectra is expressed broadening of the stretching phosphate band at 1050 cm−1, which is accomplished with the change of the intensity ratio of the peaks at 1050 and 1090 cm−1 and disappearing of the peak at 960 cm−1. All these features could be seen in the spectrum of sample A1 treated for 20 h. The observed spectral changes are in agreement with XRD data indicating changes in HAP/β-TCP: increased degree of crystalline disorder in HAP structure and partially crystalline amorphous β-TCP; and gradual decrease in the crystal sizes in both phases. The broadening of the peaks is significant after 20 h milling and is most pronounced for stretching modes of PO4 group, thus pointing for an increase in bond length distribution. The remaining sharp peaks of O–H stretching and libration modes are indicative for HAP structure preservation. The presence of a small amount of
 ions is confirmed by the weak absorption bands for A1–B20 in the range of 1410–1520 cm−1.
ions is confirmed by the weak absorption bands for A1–B20 in the range of 1410–1520 cm−1.
The formation of carbonated apatite is due to the conditions of the procedure that was carried out in air.
The position of the
 bands indicates that B type carbonate substitution dominates but small amount of OH− groups might be replaced by the CO3 groups also (Fig. 4).
bands indicates that B type carbonate substitution dominates but small amount of OH− groups might be replaced by the CO3 groups also (Fig. 4).
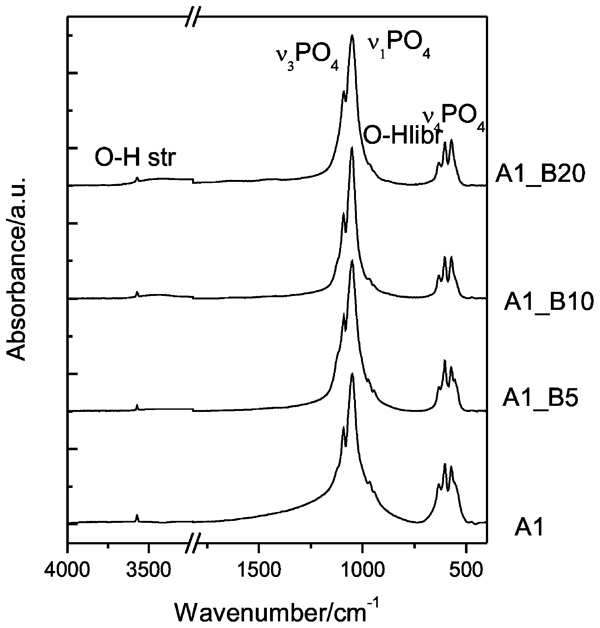
Infrared spectra of samples A1, A1–B5, A1–B10 and A1–B20
Cellular viability: Cytotoxicity test
The experimental data obtained by MTT, NR and CV assays are summarised and presented on Fig. 5. The low cell toxicity of the milled material investigated was confirmed by all of the above mentioned methods, which differ in their mechanisms and cell targets. The MTT test is based on the ability of viable mitochondria within cell to reduce succinate dehydrogenase. The basic NR dye distributes to the acidic components into the cell and therefore acts as a marker for the integrity of lysosomes and possibly of the Golgi apparatus. Crystal violet staining shows the growth rate reduction reflected by the calorimetric determination of the stained cells. No cytopathological changes were observed using double staining with acridin orange and propidium iodide. Relative cell viability was expressed as a percentage of the untreated control (100% viability) calculated for each concentration (Fig. 5).
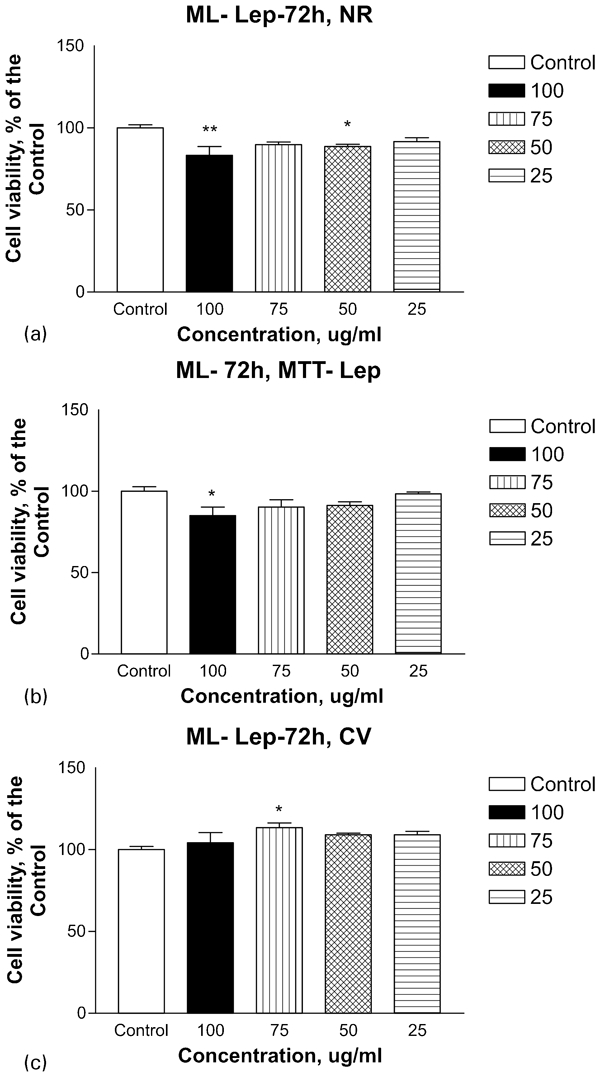
Effect of ML on viability and proliferation of cultured Lep-3 cells examined by a neutral red uptake cytotoxicity (NR) (72 h) assay, b MTT (72 h) test and c CV (72 h) staining
The present work shows that dry mechanochemical treatment of BCPs is one of the promising routes to produce nanoscale calcium phosphate materials with enhanced physicochemical properties and chemical activity. In this way, materials were obtained with alerted chemical and structural properties, controlled grain crystal sizes and in turn controlled solubility, dissolution rate and chemical activity with potential for new pronounced biological functionality.
It is desirable for the biomaterial community to understand which compositional structure parameters of a calcium phosphate phase need to be tailored in order to fine tune desired properties of material with biocompatibility.53, 54
A major objective in the field of biomaterials research is to derive synthetic nanomaterials from calcium phosphates, identical in phase compositions and structure, and nanosized crystals analogical to mineralised hard tissues. Systematic study of physical chemistry and structural characteristics of nanometric BCPs obtained after milling shows a specific nanostructural characteristics, which leads to a better understanding and control of the parameters related to the production of biomaterials with desirable and predictable 3B (bioactivity, biocompatibility and biodegradability) state. In this regard, we must mention our promising clinical results of the healing processes in treatment (with product of BCP discussed in this work) of critical size bone defects at apical periodontal lesions.55, 56 We hope that these results are encouraging for further experimental and clinical research.
In this work, it is shown that high energy milling of BCPs leads to crystalline nanosized and partially amorphous β-TCP phase differing from fully amorphous monophase TCP in analogical experimental conditions.34 This finding would be appropriate for production of biphase nanocrystalline calcium phosphates with another third part of amorphous phase. These results support the hypotheses that they are relevant to positive biological responses as a consequence of dissolution processes and effects of higher local concentration of calcium and phosphate ions. Furthermore, in the microenvironment, elevated ion concentrations may define kinetics of bone-like apatite formations in bone/implant interface and bone implant material integrations.
Conclusion
Samples of BCP sintered at 1100°C and high energy grinded for 20 h are transforming into nanosize crystalline form. The sizes are ∼36 nm for HAP and ∼33 nm for β-TCP.
The ground materials are present in the form of nanoscale particles as nanocrystals and agglomerates of few micrometres with nanostructured surfaces seen in SEM.
It was demonstrated that high energy grinding of BCPs is an easy method to obtain nanocrystallinity and agglomerates of biphase materials.
The bioactivity of the obtained materials was demonstrated by studying the viability and proliferation of (Lep) cells. It was demonstrated that the high energy ground BCP materials have a higher bioactivity. The cell survival rate in the tests was as in the control.
It is important to stress that in the BCPs β-TCP gets partially amorphous after 20 h of milling differing from the pure monophase of β-TCP, which becomes fully amorphous.
There is a real expectation that these materials have a good potential in the field of functional advanced biomaterials.
Footnotes
Acknowledgements
The authors thank the National Science Fund of Bulgaria (grant nos. DTK 02-70/2009 and DCAP-02/2/2009) for the financial support.