
Abstract
A new preceramic polymer, polyzirconosaal (PZSA), was synthesised by the ligand exchange reaction between polyzirconoxane and salicyl alcohol. The chemical structure of the precursor was analysed by Fourier transform infrared spectroscopy and 1H nuclear magnetic resonance. The precursor was air stable and exhibited excellent solubility and rheology. The thermal decomposition processes of the polymer were studied using the technique of thermogravimetric/differential thermal analysis. The thermal decomposition products of PZSA obtained in Ar atmosphere were characterised by X-ray diffraction, transmission electron microscopy and element analysis. The polymer underwent a thermal decomposition in four steps, and nanosized ZrC was formed at 1300°C via carbothermal reduction reaction of ZrO2 and carbon in argon with ceramic yield of 57·8%.
Introduction
ZrC, as one of ultrahigh temperature ceramics, combines the characteristic properties of metals and ceramics due to ionic, covalent and metallic bonding simultaneously in its NaCl type lattice structure.1–5 ZrC exhibits various outstanding physical and chemical properties, such as high hardness (∼25 GPa), high melting point (∼3420°C), high corrosion and wear resistance, solid state phase stability as well as chemical stability.6–10 Hence, ZrC has been considered a good potential material for ultrahigh temperature applications. In addition, it has been already used for making field emission arrays and for providing diffusion barrier coating on UO2 particles in nuclear industry.
ZrC particle is conventionally produced by the carbothermal reduction of carbon and ZrO2, mechanical alloying and self-propagating high temperature synthesis.11–13 However, high temperature and long production period are required because the powders are mixed on a relatively coarse scale and the synthesised products have relatively large crystallite size and poor sintering ability. Hence, hot pressing is usually necessary to produce high density bulk object. Shape and size of the products are confined and limit their broad applications. The synthesis of uniform dispersed nanosized particles has received much attention as it can increase the driving force in the sintering process and enhance the mechanical properties. Recent studies show that ZrC can be prepared by sol–gel method at low temperatures.14–16 Sacks et al. prepared ZrC and HfC powders using zirconium n-butoxide and hafnium isopropoxide, and polyhydric alcohol as the carbon sources.14 Preiss et al. used chelated derivatives of zirconium n-propoxide and various soluble carbon yielding compounds to prepare ZrC fibres, films and powders.15 Dollé et al. synthesised nanosized ZrC powders using zirconium n-propoxide and saccharose.16 The main drawback of sol–gel method is low content of ZrC in the solution, and usually the solution needs to be concentrated.
The preceramic polymer method is a relatively new and effective approach for preparing advanced ceramics by thermal decomposition of the polymers.17 The main advantages of such method of synthesising polymer derived ceramics are the applicability of polymer processing techniques, the homogeneity of the precursors on a molecular level, the low processing temperatures when compared to conventional powder sintering methods and the possibility of synthesising new compounds. Using this chemical approach, the chemistry and the polymerisation of the starting molecular precursor may be tailored to adjust the composition and the viscoelastic properties of the derived preceramic polymer and therefore prepare complex shapes (films, fibres and coatings) and ceramic matrix composites in a wide variety of non-oxide systems. Despite the numerous studies based on the precursor synthesis routes for the different classes of preceramic polymers, such as polysilanes, polysiloxanes and polysilazanes, only a few papers have focused on the synthesis of preceramic polymer for ZrC by the chemical reactions, which is the key raw material for precursor infiltration and pyrolysis (PIP) process.18, 19 Zhao et al. prepared the precursor for ZrC by just blending zirconium butoxide and divinylbenzene, but this precursor would hydrolyse when exposed to air and must be stored in inert atmosphere (nitrogen).5 Up to now, the difficulty to prepare ceramic composites by PIP process comes mainly from the lack of proper ZrC precursors.
Polyzirconoxane (PZO),20, 21 as precursors for preparing zirconia (ZrO2) fibres, is an alkane and arene soluble, water free polymer. Its ligands are quite labile in proton transfer reactions. A well known reaction of this type is the ligand exchange reaction between Zr(acac)4 and phenols in organic solvents.22 The reaction consists of proton transfer from phenols to acetylacetonate ligand, and then ligand exchange reaction with release of free acetylacetone (Hacac) occurs. Salicyl alcohol (SA) also contains phenolichydroxyl and should be able to react with PZO. In addition, SA ligand acts as carbon source to the formation of nanosized zirconium carbide during the process of carbothermal reduction reaction. Thus, in this study, we employ the ligand exchange reaction between PZO and SA to synthesise the preceramic polymers [polyzirconosaal (PZSA)] for nanosized ZrC particles. The structure of precursors was characterised by Fourier transform infrared spectroscopy (FTIR) and 1H nuclear magnetic resonance (NMR). The thermal decomposition of PZSA in inert gas atmosphere (Ar) was investigated by thermogravimetric/differential thermal analysis (TG/DTA), and the products obtained by thermal decomposition of PZSA were also characterised by X-ray diffraction (XRD), TEM and element analysis.
Experimental
Preparation of PZO
Acetylacetone (9·0 g, 0·09 mol) and triethylamine (12·2 g, 0·12 mol) were added dropwise into 260 mL methanol solution of 19·4 g (0·06 mol) zirconium oxychloride octahydrate (ZOC) below 5°C at molar ratio of Et3N/ZOC = 2·0. The reaction mixture was stirred at room temperature for 2 h and then concentrated. Addition of 150 mL tetrahydrofuran (THF) and filtration of the precipitate followed by concentration of the filtrate gave a highly viscous solution. Polyzirconoxane was isolated as a white powder by adding the viscous solution to 150 mL hexane. The molecular structure of PZO was polymers of a low degree of polymerisation with Zr–O–Zr as main chain and the ligand acetylacetone and hydroxyl group as pendants; the molecular weight was ∼2400. Fourier transform infrared spectroscopy (ν, cm−1): 3415, 1592, 1529, 1483, 1368, 1280, 1027, 932, 652, 543 and 428; 1H NMR (400 MHz, CD3OD): δ 3·6 (s, 1H, = CH– enol form), 3·1 (q, 2H, J = 8·0 Hz), 2·1 (s, 3H, CH3CO keto form), 1·4 (t, 3H, J = 8·0 Hz).
Preparation of PZSA
Polyzirconosaal was prepared by the reaction of PZO and SA. Salicyl alcohol (1·14 g) was added to the solution of 3·54 g PZO in 20 mL ethanol at room temperature with stirring. Then, the reaction mixture was stirred at reflux temperature for 4 h, and transparent orange solution was obtained. Removal of the ethanol on a rotary evaporator and drying in vacuo at 50°C for 8 h yielded a yellow solid with good solubility in common organic solvents. Fourier transform infrared spectroscopy (ν, cm−1): 3062, 2923, 2859, 1596, 1529, 1483, 1453, 1280, 1018, 752, 646 and 543; 1H NMR (400 MHz, CD3OD): δ 6·70–7·30 (m, 4H, 4×ArH), 4·6 (s, 2H, –CH2O– in phenyl ring), 3·1 (q, 2H, J = 8·0 Hz), 2·1 (s, 3H, CH3CO keto form), 1·25 (t, 3H, J = 8·0 Hz).
The dried powders were weighted in alumina boats and placed into the tubular furnace. Then, the samples were slowly heated (2°C min−1) in a sequence at 120°C (2 h), 150°C (2 h), 170°C (2 h) and 200°C (2 h) in flowing argon, and hard and brown thermosets polymers were obtained.
The obtained thermosets polymers were pyrolysed in Ar up to set temperatures. A typical heating programme was as follows: from room temperature (25°C) to set temperatures at 3°C min−1; set temperatures for 2 h; set temperatures to room temperature at 5°C min−1.
Measurements and characterisations
The raw and pretreated precursors and the samples pyrolysed at different temperatures were rubbed into powders for TG/DTA, XRD and TEM measurements. Fourier transform infrared spectra were recorded between 4000 and 400 cm−1 on a Bruker Tensor 27 spectrometer. 1H NMR spectra were recorded in methanol–d4 solvent with a Bruker AV400 spectrometer. The byproducts of the reaction were analysed by means of gas chromatography and mass spectrometry (GC/MS) (QP2010, Shimadzu, Japan). Gel permeation chromatography (GPC) analysis was performed on Waters Instrument (515 HPLC pump, 2410 refractive index detector) using THF as eluent and polystyrene as standards for calibration. Thermogravimetric analysis (TGA) was carried out on a TGA instrument (Netzsch STA 409 PC, Germany) at a heating rate of 10°C min−1 in Ar atmosphere. X-ray diffraction measurements were performed on the pyrolysed samples with a powder diffractometer (Rigaku D/M4X 2500; Rigaku Co., Japan) using a Cu Kα radiation. The morphology of the samples was examined using TEM (JEM-1011, JEOL, Japan) equipped with selected area electron diffraction. The amount of zirconium in the pyrolysed residue was determined using inductively coupled plasma emission spectrometry (Variant, Vista-MPX, USA), and the amounts of carbon and oxygen in the residue obtained by pyrolysis were measured with LECO CS-444LS and TC-436 instruments.
Results and discussion
Structure of precursor PZSA
Figure 1 shows the synthetic route of the precursor investigated in this study. The PZSA precursor was prepared by the reaction of PZO and SA in a simple and cheap chemical route. When SA was added into the solution of PZO, the mixture changed into a homogeneous solution right after stirring and the product was isolated as an air stable yellow solid. In the reaction between PZO and SA, SA deprotonation, proton transfer to acetylacetonate ligand, change of ligands and the release of free Hacac occurred. The obtained Zr containing polymer on this route served as precursors for ZrC.
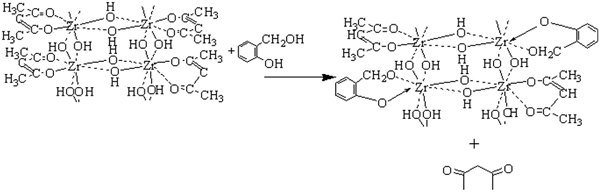
Synthetic route of PZSA precursor
There are several advantages on the used process compared with the literature methods.5, 14, 15 First, the polymer PZO was used as zirconium source, and it was stable and less toxic than zirconium alkoxides. Second, the polymer PZSA was prepared by the chemical reaction between PZO and SA, in which Zr element dispersed at molecular level, not just by mixing of the materials. Last, when the precursor was exposed to air, it would not hydrolyse, so it was air stable. This precursor exhibited excellent solubility in common organic solvents, such as methanol, ethanol, THF and CHCl3; for example, 1 g ethanol can dissolve up to 0·8 g PZSA. Furthermore, when this precursor was dissolved in ethanol, the viscosity of this solution can be tailored in the range of 200–400 mPa s at 25°C and therefore can be used to prepare C/C–ZrC composites via PIP process.
The FTIR and 1H NMR spectra of PZSA were shown in Figure 2 Figs. 2 and 3 respectively. As shown in Fig. 2, the strong absorption at 752 cm−1 was due to C–H of aromatic in SA ligand, absorption at 1596 and 1529 cm−1 was due to C = O and C = C in acetylacetonate ligand, and absorption at 543 cm−1 was due to Zr–O–Zr. The occurrence of chemical reactions was confirmed by the fact that obvious decrease in intensity of 1018 cm−1 was due to methylol groups in SA and the decrease in the intensity of 1529 cm−1 assigned to C = C in acetylacetonate ligand. In Fig. 3, 1H NMR spectrum showed multiplets and broadened peaks, as is expected in the polymer structure. A triplet at 1·2 ppm was due to CH3CO enol form, a broad singlet at 2·1 ppm was due to CH3CO keto form, the quartet at 3·1 ppm was due to CH2 keto form; a singlet at 4·6 ppm was due to CH2O in SA ligand; and signals at 6·70–7·30 ppm were due to aromatic protons.
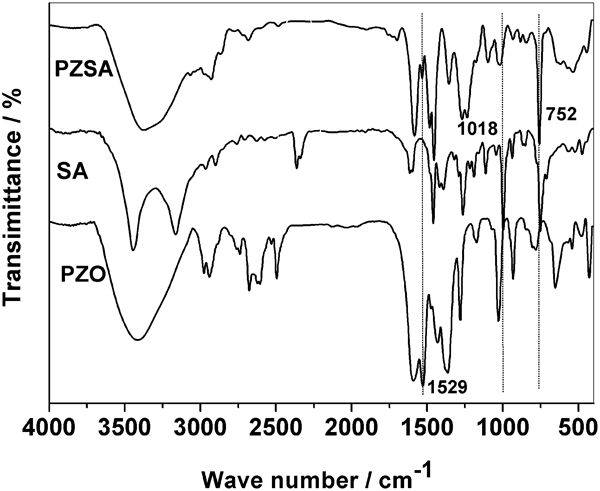
Fourier transform infrared spectra of PZO, SA and PZSA
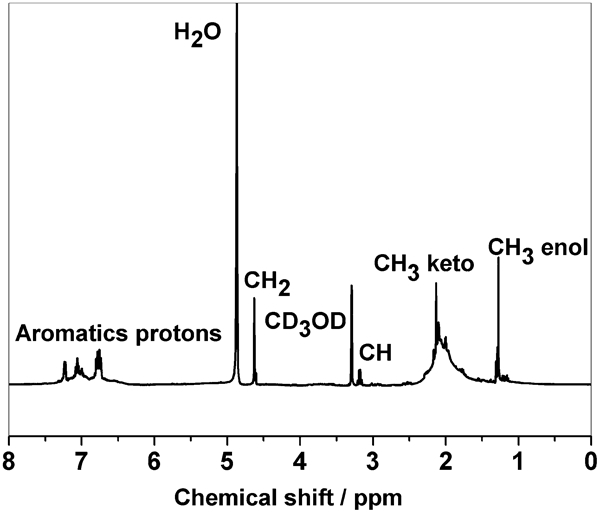
1H NMR spectrum of PZSA
Number average molecular weights in the range of 2000–3000 relative to polystyrene standards were determined by GPC. It was worth noting that a higher molecular weight precursor was obtained by slowly heating the PZSA to 200°C in flowing argon. Then, PZSA became not soluble in organic solvents or melts after cure treatment. For the linear oligomer turned into the three-dimensional network and a crosslinked polymer, PZSA was formed.
Free acetylacetone was found by GC/MS analysis on the distilled solvent, indicating that partial free acetylacetone was released in the solvents. The proposed reaction mechanism is that SA deprotonates and a proton transfers to acetylacetonate ligand, resulting in the partial release of acetylacetone and chelating of SA to central zirconium atom. According to the results of FTIR, 1H NMR, GPC and GC/MS, the ligand exchange reaction between PZO and SA was confirmed, and the obtained PZSA might be a linear Zr–O–Zr chain polymer, with acetylacetonate and deprotonated SA as ligands, and the molecular weight is about 2000–3000.
Thermal behaviour
To study the polymer to ceramic conversion of the precursor, a series of pyrolysis trials were conducted in tube furnace and TG experiments. Figure 4 depicted the typical TG/DTA curve of the precursor after cross-linking treatment at 200°C. The TGA curve showed four-stage weight loss events: 25–200°C, 200–500°C, 500–1100°C and 1150–1375°C. There was no apparent weight loss from 25 to 200°C. The considerable weight loss (30%) between 200 and 500°C was due to the decomposition of organic groups. Accordingly, DTA curve showed a sharp endothermic peak at 479·5°C, and no further weight loss was detected between 500 and 1100°C. After 1100°C, the weight loss accelerates again and does not stabilise up to 1375°C, which is due to the carbothermal reduction between ZrO2 and carbon as shown below. The ceramic yield was 53·5% at 1375°C, and the residue was black
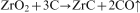
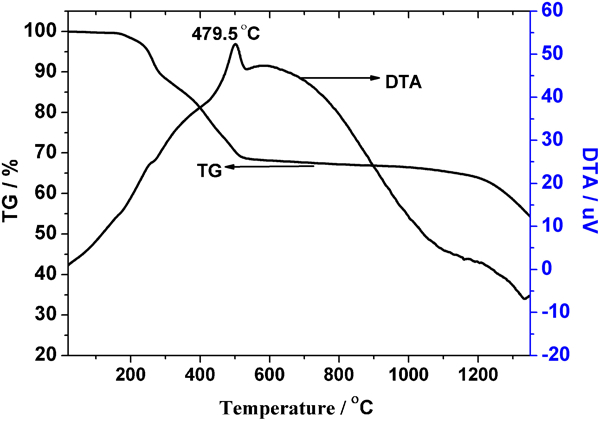
Thermogravimetric/differential thermal analysis curve of PZSA in Ar with heating ratio of 10°C min−1
X-ray diffraction patterns of ceramics
X-ray diffraction results of the precursor powders at different heat treatment temperatures were shown in Fig. 5. As can be seen, there was no diffraction peaks observed in the curve of 400°C, which meant that the products were amorphous and did not crystallise. The onset of tetragonal ZrO2 (t-ZrO2) crystallisation was apparent by XRD in the 500°C products, but the peaks were broad. With the temperature increase from 600 to 1000°C, t-ZrO2 was the only crystalline phase and the only significant change in the XRD pattern was that the t-ZrO2 peaks increased in intensity. There was no diffraction peak of carbon in the curves because carbon remained amorphous. Upon heat treatment at 1100°C, some t-ZrO2 had transformed to monoclinic ZrO2 (m-ZrO2), and the initial formation of ZrC was observed at 1100°C, which was much lower than the temperature 1653°C by thermodynamics calculation.23 This was because carbon and zirconium oxide had been reacted at the molecular level, and pyrocarbon was produced in situ and was more active. According to the Zr–C phase diagram,4 ZrC phase is a solid solution with wide carbon stability range (about 38–50 at-%). Thus, different ZrC phases will be formed with changed temperature or C/Zr ratio. At 1200°C, the ZrC peaks increased in intensity. At 1300°C, the XRD pattern showed characteristic peaks at 2θ = 33·1° (111), 38·4° (200), 55·4° (220), 65·9° (311) and 69·3° (222), which corresponded to the typical structure ZrC diffraction (Joint Committee on Powder Diffraction Standards no. 35-0784).2, 14, 19 Sharp diffraction peaks indicated good crystallinity of ZrC nanoparticles, and no characteristic peak related to any impurity was observed. The average ZrC crystallite size is calculated by Scherrer's formula using the (111), (200), (220), (311) and (222) reflections of ZrC crystal planes. However, TGA and element analysis results showed that the 1300°C sample was not phase pure stoichiometric ZrC. Instead, the sample consisted of ‘zirconium oxycarbide’ (i.e. ZrC with some oxygen dissolved in lattice) and some residual amorphous carbon.
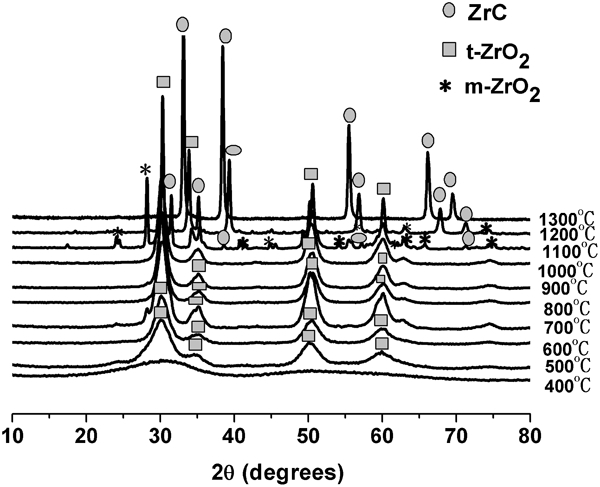
X-ray diffraction patterns of ceramic products obtained at various temperatures
Element contents of ceramics
Table 1 lists the element contents of the synthesised ZrC powders at different heat treatment temperatures for 2 h. It can be seen that carbon and oxygen contents decrease with increasing temperature. A sample pyrolysed at 1100°C had a carbon content of 22·5 wt-%. Table 1 also shows that subsequent heat treatment at 1300°C produces a sample with 16·3 wt-% carbon. This carbon content is greater than the expected value of 11·6 wt-% for phase pure stoichiometric ZrC. Hence, it is apparent that the sample retains some free carbon after the heat treatment. Furthermore, the sample also contains 4·8 wt-% oxygen. Since ZrO2 is not observed in XRD patterns for samples heat treated at or above 1300°C, it is evident that the oxygen present in the sample is dissolved in the zirconium carbide lattice. Table 1 also shows that further heat treatment at 1400°C produces a sample with carbon content 14·8 wt-% and the oxygen content was reduced to a low level. The decrease in carbon and oxygen contents for the samples is consistent with the weight loss behaviour in TGA curve (Fig. 4) above the temperature of 1200°C. In combination, the weight loss and the decrease in carbon and oxygen contents upon heat treatment up to 1400°C can be explained by a carbothermal reduction reaction.
Element contents of synthesised ZrC powders at different heat treatment temperatures/wt-%

Transmission electron microscopy images of ceramics
Figure 6 shows the TEM images of the synthesised ZrC powders at 1300°C for 2 h with selected area electron diffraction analysis. The crystallite sizes of the synthesised powders mainly distribute over 50–100 nm but exhibit agglomerated particle morphology. There are some floccules under or around the ZrC particles, and this is believed to be amorphous carbon. The selected area electron diffraction pattern shows the diffraction of a certain ZrC particle that is well crystallised.

Transmission electron microscopy and selected area electron diffraction image of synthesised ceramics at 1300°C for 2 h
Conclusion
In summary, ZrC nanoparticles have been successfully synthesised using a new precursor via preceramic polymer method. Using PZO and SA as precursors can produce air stable PZSA with excellent solubility and rheological properties. The thermal decomposition of PZSA precursors consisted of four stages, such as decomposition of organic residues and carbothermal reduction reaction between ZrO2 and carbon. Pure ZrC phase is formed at 1300°C/2 h by pyrolysis of PZSA in an argon atmosphere. The synthesised ZrC distributes over 50–100 nm with agglomerated particle morphology evidenced by the results of TEM and XRD respectively. The synthesised nanoparticles are expected to be suitable for the pressureless sintering of ZrC ceramic, and this method can be extended to the synthesis of other metal carbides and borides.
Footnotes
Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (grant nos. 2011QNA20 and 2012DXS02) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.