
Abstract
The extent to which the composition of the reaction mix affects the formation of a biphasic C–(A)–S–H/N–A–S–H geopolymer framework, and how the interaction of these phases affects geopolymer microstructure, can only be studied by strict stoichiometric control. Stoichiometrically controlled geopolymers containing both C–(A)–S–H and N–A–S–H gels are produced here by reaction of a sodium silicate solution with calcium aluminosilicate powders, which were synthesised via a novel solution-polymerisation method utilising polyethylene glycol as a polymer carrier to sterically inhibit movement of precursor cations. Increased Ca content in the reaction mix appears to promote greater formation of a C–(N)–(A)–S–H gel, while reduced Ca content and increased Al and Si content promote greater formation of an N–A–S–H gel in addition to the main C–(A)–S–H reaction product. The stoichiometrically controlled geopolymers constitute a chemically simplified model system through which the nature of the biphasic C–(A)–S–H/N–A–S–H gels present in alkali activated binders can be studied.
Introduction
Geopolymers and alkali-activated binders offer a viable low CO2 alternative to ordinary Portland cement and exhibit desirable technical properties as well as a potential reduction in CO2 emissions by over 80%.1–3 Industrial byproducts such as fly ash and granulated blast furnace slag are often used as raw materials for the production of geopolymers and alkali-activated binders, and the chemical composition of these materials varies significantly between sources.2,4 Variations in the composition of the raw material can significantly affect the technical properties exhibited by these binders, and as such, there is currently no single formulation that can be used to produce geopolymers and alkali-activated binders that possess similar physical and chemical characteristics across a wide range of precursor compositions.2,4
Coexistence of both C–(A)–S–H and N–A–S–H gel frameworks within alkali-activated binders results in complex thermodynamic and chemical interactions, which dictate material properties and performance. Despite the vast number of studies investigating the chemistry of alkali-activated binders, the literature is often conflicting, and the experimental analysis involves a large number of unconstrained parameters. This is, at least in part, due to the production of alkali-activated binders using raw materials that display large variations in chemical and mineralogical composition. It is therefore necessary to develop a method to study alkali-activated binders with strict control of the stoichiometry in these systems to gain a more complete understanding of the relationships between thermodynamics, reaction kinetics and the initial compositions of the precursors used to produce alkali-activated materials.
Sol–gel based procedures 5 have been used to synthesise and investigate the effect of composition on C–(A)–S–H and N–A–S–H phase assemblages6–10 in the past, as well as to conduct compatibility studies of C–S–H and N–A–S–H gels.8,10 However, these studies mostly investigate the effect of addition of alkali cations, alkaline earth cations or aluminium on the properties of C–S–H or N–A–S–H gels after formation of the gel. In order to accurately represent the physical and chemical interactions occurring during the actual process of alkali-activation, all of the ionic species of interest must be present in a precursor powder prior to alkali-activation.
This study examines the effect of precursor material composition on C–(A)–S–H and N–A–S–H phase assemblage by synthesising calcium, silicon and aluminium containing mixed oxides via an organic polymeric steric entrapment solution-polymerisation route. 11 This method utilises polyethylene glycol (PEG) to sterically inhibit the movement of metal cations during solution-polymerisation, forming a homogeneous mixed oxide powder upon drying. The stoichiometrically controlled reactive precursor powders are subsequently activated with an alkaline solution to form an alkali-activated binder. The effects of Ca, Si and Al in the reaction mix on the chemistry and microstructure of the geopolymer binder are assessed here by electron microscopy, X-ray diffraction (XRD) and nuclear magnetic resonance (NMR) spectroscopy.
Experimental
Precursor powder synthesis
A 5 wt-% PEG solution was made by adding PEG powder (Sigma Aldrich, molecular weight 20 kDa) to distilled water and stirring at 60°C. Aluminium nitrate nonahydrate, Al(NO3)3.9H2O (Sigma Aldrich, 98.5 wt-%), and calcium nitrate tetrahydrate, Ca(NO3)2.4H2O (Sigma Aldrich), were added to distilled water to produce 40 wt-% solutions of each, which were then added to the 5 wt-% solution of PEG and stirred at 60°C. Colloidal silica, SiO2 (Sigma Aldrich, 40 wt-% in water), was added to the 5 wt-% solution of PEG containing aqueous aluminium and calcium nitrates, and water was evaporated by stirring over heat at 80°C to form a viscous aerated gel. The stoichiometry of the metal cation PEG solution was designed so that the ratio of positive valences from the metal cations to negative valences from the polymer was 2.0. The viscous aerated gel was then placed in a drying oven at 100°C overnight. The dry polymer matrix was then calcined at 900°C to produce a fine white powder, which was subsequently ground by hand using a mortar and pestle in preparation for characterisation and alkali-activated binder synthesis.
Alkali activated binder synthesis
The activating solution was prepared by dissolution of sodium hydroxide powder (AnalaR, 99 wt-%) in sodium silicate solution (N grade, 37.5 wt-%, PQ Australia) and distilled water. The reaction mixtures had an activating solution modulus of SiO2/Na2O = 1, a water/solids ratio of 0.75 and cation ratios as outlined in Table 1. The activating solution was mixed with the precursor powder to form a homogeneous paste, which was subsequently cast in sealed containers and cured at ambient temperature for 3 and 28 days.
Molar ratios of the reaction mix for each sample

Characterisation
X-ray diffraction data were collected using a Bruker D8 Advance instrument with Cu Kα radiation, a nickel filter, a step size of 0.020° and a counting time of 3 s/step. Environmental scanning electron microscopy (ESEM) was conducted using an FEI Quanta instrument with a 15 kV accelerating voltage, a working distance of 10.0 mm and a Link-Isis (Oxford) X-ray energy dispersive (EDX) detector. 29Si magic angle spinning (MAS) NMR spectra were collected at 119.141 MHz on an Agilent (Varian) VNMRS-600 (14.1 T) instrument with a pulse width of 7 μs, a relaxation delay of 120 s and 1024 scans. 27Al MAS NMR spectra were acquired on the same instrument at 156.261 MHz, with a pulse width of 4 μs, a relaxation delay of 2 s and 1024 scans.
Results and discussion
X-ray diffraction
X-ray diffractograms of the calcined precursor powder and the alkali-activated binders are presented in Fig. 1. The X-ray diffractograms of the calcined precursor powders display two broad, featureless humps centred at ∼14° 2θ and ∼30° 2θ, indicating a predominantly amorphous material. Small amounts of tricalcium aluminate (C3A; PDF # 33-0251) and two polymorphs of dicalcium silicate (C2S; PDF # 33-0302 and 36-0642) are identified in the calcined precursor powder of both samples, while a small amount of crystalline calcium oxide (PDF # 48-1467) is also identified in the calcined precursor powder of sample B. Calcination of the precursor powder to 900°C is necessary to remove any calcium carbonate present in the sample (converting it to free lime); however, it is evident from the X-ray diffractograms in Fig. 1 that the calcination process has caused some devitrification of the amorphous material and subsequent formation of a small amount of these crystalline phases. Alkali activation of both samples A and B produces a much broader amorphous hump centred at approximately 29° 2θ, characteristic of alkali activated materials and indicative of the formation of a predominantly amorphous reaction product consistent with that formed during alkali-activation of granulated blast furnace slag.12,13 The main reaction product in both samples is a poorly crystalline aluminium substituted calcium silicate hydrate (C–(A)–S–H) phase with structural similarity to aluminium containing tobermorite (PDF # 19-0052).
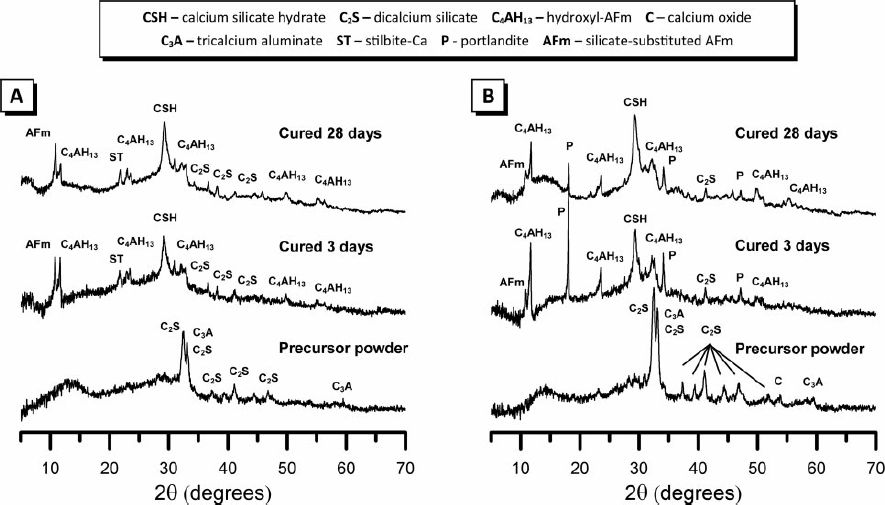
X-ray diffractograms of calcined precursor powder and alkali activated binders cured for 3 and 28 days for samples A and B as marked
In both samples, the crystalline phases present in the calcined precursor powder are partially consumed during alkali-activation, leading to the formation of poorly ordered C–(A)–S–H and crystalline C4AH13 (PDF # 02-0077). C2S is progressively consumed as the alkali-activation reaction continues. Small amounts of stilbite Ca (NaCa2Al5Si13O36.14H2O) (PDF # 44-1479) are evident in the alkali-activated binders cured for 3 and 28 days for sample A, while the diffractograms of the alkali-activated binder cured for 3 and 28 days for sample B indicate the presence of portlandite (Ca(OH)2) (PDF # 44-1481), which is likely to have, in part, formed as a result of dissolution and hydration of C2S. A partially silicate substituted AFm phase [Ca2Al(OH)6]·X·xH2O, where X is a doubly charged carbonate or aluminosilicate anion) is also observed in both samples.14,15 Increased calcium content in the precursor powder appears to promote the formation of portlandite in addition to the main C–(A)–S–H and C4AH13 products.
Scanning electron microscopy
The compositions of the alkali-activated binders for samples A and B as determined by ESEM–EDX are reported in Fig. 2. The chemistry of both alkali-activated binders differs from that of the reaction mix. The Ca/(Al+Si) ratio of the alkali-activated binder for sample A is lower than that of the reaction mix, while the alkali-activated binder of sample B is more Ca rich and Si deficient than the reaction mix, with a slight increase in Al content. It is also clear that Na has been incorporated into the binder for both samples, likely assuming a charge balancing function within a sodium and aluminium substituted calcium silicate hydrate (C–(N)–(A)–S–H) gel. Values for the Ca/(Al+Si) ratio exhibited by the alkali-activated binder for sample B lie along a line that can be drawn between the Ca/(Al+Si) ratios of a C–(A)–S–H gel and a partially silicate substituted AFm phase. This is likely to be a result of intermixing of the C–(A)–S–H gel and AFm and portlandite phases. The lower Ca content in the reaction mix for sample A seems to promote the formation of a C–(N)–(A)–S–H gel, whereas the higher content of Ca in the reaction mix for sample B appears to promote the formation of a C–(A)–S–H gel. 10
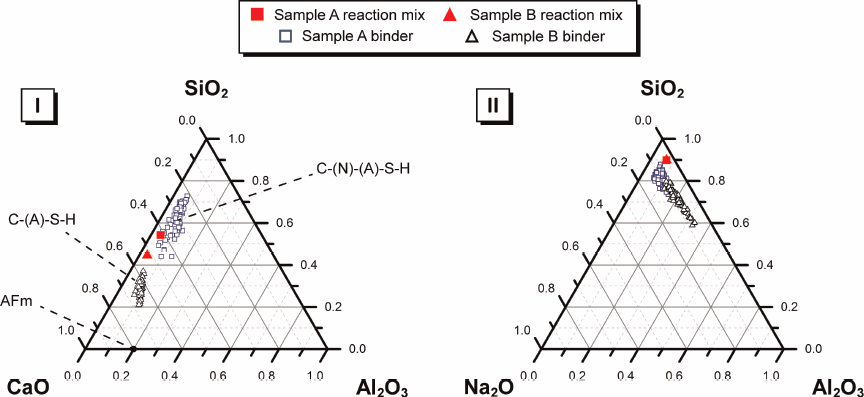
Projection of alkali activated binder chemistry onto (I) ternary CaO–Al2O3–SiO2 system (neglecting Na2O content) and (II) ternary Na2O–Al2O3–SiO2 system (neglecting CaO content) showing elemental composition of alkali activated binders cured for 3 days for samples A and B as determined by ESEM-EDX analysis
The compositional range for single phase C–(A)–S–H, as determined by experimental observation16,17 and thermodynamic modeling, 18 is less extensive than the values observed for the alkali-activated binders in Fig. 2. As such, it is likely that the chemistry of the alkali-activated binders for samples A and B is a combination of the chemistry of both highly crosslinked framework silicate (N–A–S–H) and chain silicate (C–(N)–(A)–S–H or C–A–S–H) gel structures, as well as contributions from the portlandite and AFm phases identified by XRD. This suggests that the lower Ca content in the reaction mix for sample A promotes the formation of a sodium aluminium silicate hydrate (N–A–S–H) gel in addition to the C–(N)–(A)–S–H gel, whereas the higher content of Ca in the reaction mix for sample B promotes greater inclusion of Al in the C–(N)–(A)–S–H binder. The main reaction product will be a mixture of C–(N)–(A)–S–H and N–A–S–H gels 18 ; however, lower levels of Ca in the reaction mix will drive the substitution of Al to form a C–A–S–H gel, as well as incorporation of Na to form a C–(N)–(A)–S–H gel, and this will progress to a point that achieves the maximum Al and Na incorporation that is thermodynamically stable. The decreased Ca/(Al+Si) ratio in the reaction mix of sample A will drive increased formation of N–A–S–H. Higher concentrations of Ca in the initial reaction mix (as is the case for sample B) will drive further development of C–(N)–(A)–S–H.
Nuclear magnetic resonance spectroscopy
The 27Al MAS NMR spectra for the calcined precursor powders and alkali-activated binders are presented in Fig. 3. The spectra of the two calcined precursor powders are very similar, displaying a broad tetrahedral Al resonance centred at approximately 54 and 56 ppm for samples A and B respectively. The breadth of this resonance indicates that there is a distribution of Al environments in each sample, rather than a single well defined site. This resonance is attributed to the glassy phase responsible for the broad amorphous hump in the X-ray diffractograms of the uncalcined precursor powders and is similar to that observed for granulated blast furnace slag.12,13 Both samples A and B display a low intensity broad resonance centred at approximately 0 ppm, partly overlapping the downfield spinning side band of the main Al(IV) peak, attributed to octahedral Al. This is consistent with the small amount of C3A identified by XRD in both calcined precursor powders.
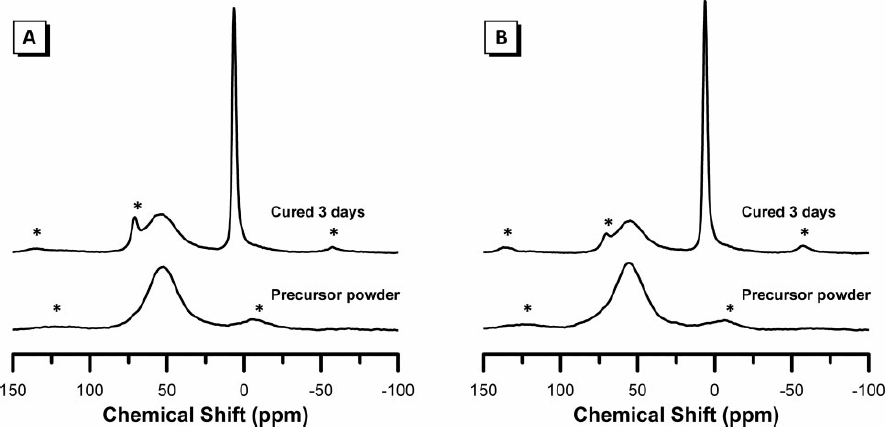
27Al MAS NMR spectra of (A) sample A and (B) sample B: expected position of spinning side bands are indicated by*
The 27Al MAS NMR spectra of the alkali-activated binders for both samples A and B display a low intensity broad resonance from 30 to 70 ppm centred at approximately 55 ppm and again assigned to Al in a significantly distorted tetrahedral environment, as well as a low intensity narrow resonance at approximately 72 ppm assigned to Al in a well defined tetrahedral coordination and a high intensity narrow resonance at approximately 6.5 ppm assigned to Al in a well defined octahedral coordination. A small contribution to the resonance at 72 ppm is also expected from a spinning side band of the sharp resonance at 6.5 ppm, as indicated in Fig.3. Ordered C–(A)–S–H identified in the alkali-activated binders for both samples by XRD will contain Al substituted for Si in a tetrahedrally coordinated environment, leading to the well defined resonance observed at 72 ppm, while poorly crystalline C–(A)–S–H and N–A–S–H will each contain Al in significantly distorted tetrahedral environments, leading to the broad resonance observed between 30 and 70 ppm and centred at approximately 55 ppm. The secondary reaction product C4AH13 is responsible for the well defined resonance observed at 6.5 ppm. 19
The 29Si MAS NMR spectra of the calcined precursor powders and alkali-activated binders are presented in Fig. 4. For both samples A and B, the spectra of the calcined aluminosilicate powder display a broad resonance centred at approximately − 82 ppm. Due to its broad nature, it is likely that this resonance contains contributions from each of the Q1, Q2 and Q2(1Al) lower connectivity silicon environments as well as the highly Al substituted Q4 silicon coordination environments. 12 Alkali-activation of the calcined precursor powder for sample A causes a significant reduction in the intensity of the Q1 region of the spectra and a significant increase in intensity at − 81 and − 84 ppm. This region is assigned to Q2 and Q2(1Al) environments, indicating the formation of a C–(A)–S–H gel. 12 A significant increase in intensity is observed within the − 90 to − 100 ppm region of the spectra commonly assigned to aluminium substituted Q3 and Q4 species, suggesting the formation of a N–A–S–H gel, with some contribution to this resonance expected from the aluminosilicate anion present in the AFm secondary reaction product as observed by XRD.
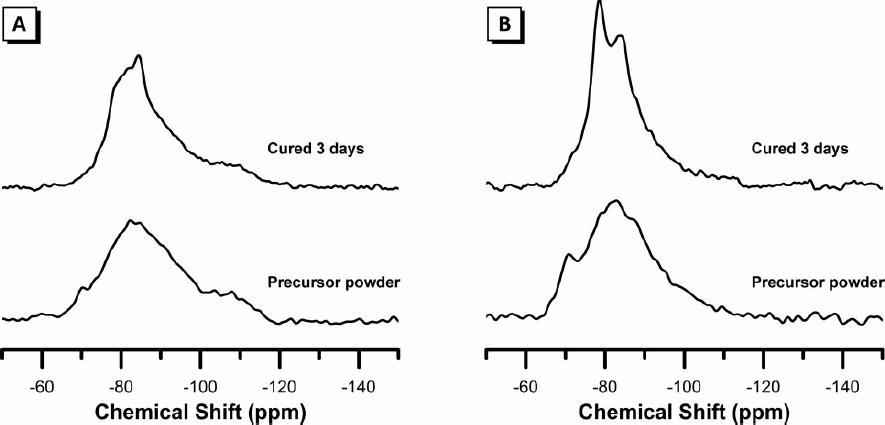
29Si MAS NMR spectra of (A) sample A and (B) sample B
Similarly, upon alkali-activation, sample B displays a significant reduction in the intensity of the Q1 region of the spectra, as well as a slight reduction in intensity in the Al substituted Q4 region of the spectra, and a significant increase in the intensity at − 81 and − 84 ppm. This increase is again assigned to the formation of Q2 and Q2(1Al) environments present in a C–(A)–S–H gel. The increased intensity of the resonance assigned to Q3 species upon alkali-activation of sample A suggests that a lower Ca content in the reaction mix promotes the formation of a N–A–S–H gel in addition to the main C–(A)–S–H reaction product, while the higher Ca content in the reaction mix promotes greater formation of the C–(A)–S–H reaction product. This is in agreement with the ESEM-EDX observations (Fig. 2) that the alkali activated binder of sample B is more Al rich than the bulk chemistry of the reaction mix.
Conclusions
Stoichiometrically controlled alkali-activated binders were synthesised via alkali-activation of calcium aluminosilicate precursor powders. These precursor powders were synthesised from aqueous precursor solutions via an organic steric entrapment solution-polymerisation route utilising PEG to sterically inhibit the movement of precursor cations. The main reaction product in both samples was a biphasic C–(A)–S–H/N–A–S–H gel. Increased Ca content within the reaction mix appears to promote greater formation of C–(N)–(A)–S–H, while reduced Ca content and increased Al and Si content in the reaction mix appear to promote greater formation of N–A–S–H in addition to the main C–(A)–S–H reaction product. The stoichiometrically controlled alkali-activated binders constitute a chemically simplified model system through which the nature of the biphasic C–(A)–S–H/N–A–S–H gels present in alkali activated binders can be studied.
Footnotes
Acknowledgements
This work was funded in part by the Australian Research Council (ARC), including support through the Particulate Fluids Processing Centre, a Special Research Centre of the ARC, and through an Australian Postgraduate Award supporting the doctoral studies of BW.