
Abstract
Favorable effects of angiotensin II type 1 receptor blockers on patients with ischemic or idiopathic dilated cardiomyopathy (DCM) have already been suggested by several human trials, but their effects on DCM remain unknown. Hence, we investigated the effect of olmesartan on myocardial remodeling in a rat model in which myosin-induced experimental autoimmune myocarditis (EAM) might develop into DCM. EAM was elicited in Lewis rats by immunization with cardiac myosin, and 28 d after immunization, the surviving Lewis rats were divided into two groups and treated with either olmesartan (10 mg/kg/d) or vehicle. Age-matched normal rats without immunizations were also used. After four weeks of treatment, we investigated the effects of olmesartan on cardiac function, inflammatory cytokines and cardiac remodeling in EAM rats. Myocardial functional parameters measured by hemodynamic and echocardiographic analyses were significantly improved by the treatment with olmesartan compared with those of vehicle-treated rats. Olmesartan significantly reduced cardiac fibrosis as well as hypertrophy and its molecular markers (left ventricular [LV] mRNA expressions of transforming growth factor beta1, collagen-I and -III, and atrial natriuretic peptide) compared with those of vehicle-treated rats. Increased myocardial mRNA expressions of proinflammatory cytokines (interleukin [IL]-6, IL-1β), monocyte chemoattractant protein-1 and matrix metalloproteinases (MMP-2 and -9) were also suppressed by the treatment with olmesartan in rats with DCM. Further, the plasma level of angiotensin II was significantly increased in olmesartan-treated rats. These findings demonstrate that olmesartan treatment significantly improved LV function and ameliorated the progression of cardiac remodeling in rats with DCM after EAM.
Introduction
Myocarditis often progresses to dilated cardiomyopathy (DCM), a major cause of heart failure. In our study, we used a rat model of myosin-induced experimental autoimmune myocarditis (EAM), in which the heart transits from an acute phase (inflammatory myocarditis) to a chronic phase (remodeling and DCM). EAM of rats mimics human idiopathic giant cell myocarditis. 1,2 DCM is recognized as a significant cause of morbidity and mortality. Treatment strategies of DCM vary owing to its diverse etiopathology ranging from myocardial infarction to myocarditis (bacterial, viral, parasitic and autoimmune) among others.
A growing body of evidence shows that angiotensin II (Ang-II) type 1 receptor (AT1R) blockers inhibit cardiac hypertrophy and remodeling, and prevent progression of systolic heart failure, thereby reducing cardiac morbidity and mortality; 3,4 they also reduce myocardial damage during myocarditis. 5 The transition from compensated to failing cardiac hypertrophy has been attributed to a reversal to fetal pattern cardiomyocyte gene expression and results in ventricular remodeling. 6,7 Many kinds of cytokines, such as basic fibroblast growth factor, Ang-II, transforming growth factor beta1 (TGF-β1) and collagen-III, have been suggested to play an important role in structural remodeling of the non-myocyte compartment of the myocardium following heart failure. 8,9
The major cardiovascular actions of Ang-II have been reported to be mediated by the AT1R, and AT1R antagonists are therapeutically effective for the treatment of patients with heart failure by reducing cytokines and oxidative stress through its anti-inflammatory effects. 10–12 Thus, the blockade of AT1Rs is another way to interrupt the renin–angiotensin system (RAS). Recently, olmesartan, an AT1R antagonist, has been shown to ameliorate EAM by the suppression of myocardial damage and inflammatory events in the myocardium in addition to hemodynamic modifications, 5,13 and it has also been reported to inhibit nitric oxide (NO) production in macrophages 14 and interleukin (IL)-1β production. 13,15 We previously reported that candesartan treatment decreased myocardial fibrosis and its marker molecules (i.e. mRNA expression of TGF-β1 and collagen-III), and improved the survival rate and cardiac function in rats with DCM after myocarditis in a dose-dependent manner. 16 Furthermore, it has been reported that valsartan, another drug in this class, suppressed myocardial hypertrophy and fibrosis, and improved the hemodynamics and cardiac function in an animal model of postmyocarditis DCM. 17 A significant number of reports have accumulated concerning the effects of AT1R blockers on myocardial infarction and myocardial hypertrophy using animal models. So far, only a few studies have revealed the effects of AT1R blockade in DCM after EAM, 16,17 and moreover, studies have not been assessed for the effects of AT1R blocker on inflammatory cytokines and ECM remodeling in rats with DCM after EAM.
EAM was demonstrated to progress into the clinicopathological state similar to DCM in the chronic phase, and was found to be characterized by the enlargement of the heart, dilation of ventricles, diffuse and extensive myocardial fibrosis, and hypertrophic and atrophic changes of myocardial fibers, resembling human cardiomyopathy. 2,18 However, the effects of olmesartan has been investigated acutely in EAM rats, 5,13 but has not been explored chronically (i.e. DCM). Thus, the aim of this study was to clarify whether AT1R antagonist would have beneficial effects on cardiac function, proinflammatory cytokines and cardiac remodeling in a rat model of DCM after EAM.
Materials and methods
Materials
Olmesartan was generously provided by Daichi-Sankyo Pharmaceutical (Tokyo, Japan), and Lewis rats (male, 8 weeks old) were purchased from Charles River Japan Inc (Kanagawa, Japan).
Experimental design
All experiments were carried out using eight-week-old male Lewis rats and were performed in accordance with the guidelines of our institute. 19 Lewis rats were injected in the footpads with antigen-adjuvant emulsion in accordance with a procedure described previously. 1,19 In brief, porcine cardiac myosin was dissolved in phosphate-buffered saline at 5 mg/mL and emulsified with an equal volume of complete Freund's adjuvant with 11 mg/mL Mycobacterium tuberculosis H37RA (Difco Lab., Detroit, MI, USA). EAM in rats was induced by immunization with 0.1 mL of emulsion once by subcutaneous injection into the rear footpads (0.1 mL to each footpad). The morbidity of EAM was 100% in rats immunized by this procedure. 1,19 Twenty-eight days after immunization, the surviving Lewis rats were divided into two groups and received oral administration of olmesartan (10 mg/kg/d; Group Olm-10) or vehicle (Group V) for 28 days. Age-matched Lewis rats without immunization were used as normal controls (Group N). Since fibrosis plays an important role in left ventricular (LV) remodeling in our model, we have chosen the dose of olmesartan (10 mg/kg) on the basis of antihypertensive and antifibrotic properties demonstrated in earlier reports. 5,13,20
Hemodynamic and echocardiographic studies
To obtain hemodynamic data, rats were anesthetized with 2% halothane in oxygen during the surgical procedures. A catheter-tip transducer (Miller SPR 249; Miller Instruments, Houston, TX, USA) was introduced into the left ventricle through the right carotid artery for the determination of peak LV pressure (LVP) and LV end-diastolic pressure (LVEDP), and the rates of intraventricular pressure rise (+dP/dt) and decline (−dP/dt) were recorded as described previously. 16 After instrumentation, the concentration of halothane was reduced to 0.5% to minimize the anesthetic effect on hemodynamic parameters. Echocardiographic studies were carried out with a 7.5-MHz transducer (Aloka Inc, Tokyo, Japan). The LV dimensions in diastole (LVDd) and systole (LVDs) and percentage fractional shortening (FS) were estimated using M-mode measurements.
Histopathology
The body weight (BW) of rats was noted just before the surgical procedure. After the hemodynamic and echocardiographic analyses, the rats were killed, and the wet myocardium was isolated and weighed to calculate the ratio of heart weight (HW) to BW. The excised myocardium was kept in formalin and the mid-ventricle sections were then embedded with paraffin. The area of myocardial fibrosis in LV tissue sections stained with Azan–Mallory was quantified using a color image analyzer (CIA-102, Olympus, Tokyo, Japan) and measured the blue fibrotic areas as opposed to the red myocardium at ×200 magnification. The results were presented as the ratio of the fibrotic area to the whole area of the myocardium. 15
Using hematoxylin and eosin (H&E) sections, myocyte diameter measurements were performed in 10 myocytes selected per field in 400-fold magnification by light microscopy. Short axis diameters of each myocyte were measured from the hearts of all groups of rats. Each average value was obtained from the data for 10 myocytes and was used as independent sampling data. In addition, inflammatory cell infiltrations were identified using H&E-stained sections at 100-fold magnification by light microscopy.
Estimation of plasma and tissue Ang-II by radioimmunoassay
Blood samples were collected by heart puncture immediately after echocardiographic measurements, and were transferred into a chilled glass tube containing 0.25 mL of 125 mmol/L EDTA and 25 mmol/L o-phenanthroline for the purpose of subsequent determinations of plasma Ang-II by standardized radioimmunoassay (RIA). 21 The extraction of tissue Ang-II was performed in accordance with the method of Phillips and Stenstrom. 22 Frozen tissues were homogenized in 10 volumes of 1 N acetic acid containing 10 μg/mL pepstatin and concentrated by passing through C-18 Sep-Pak cartridges (Waters Corporation, Milford, MA, USA). Ang-II concentrations were determined by RIA. 21
Immunohistochemical assay
Formalin-fixed, paraffin-embedded cardiac tissue sections were used for immunohistochemical staining. After deparaffinization and hydration, the slides were washed in Tris-buffered saline (TBS; 10 mmol/L Tris HCl, 0.85% NaCl, pH 7.5) containing 0.1% bovine serum albumin. Endogenous peroxidase activity was quenched by incubating the slides in methanol and 0.6% H2O2 in methanol. To perform antigen retrieval, the sections were pretreated with trypsin for 15 min at 37°C. After overnight incubation with the primary antibody, namely, mouse polyclonal anti-inducible NOS (iNOS) antibody (diluted 1:100) (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) at 4°C, the slides were washed in TBS and horseradish peroxidase-conjugated secondary antibody was then added and the slides were further incubated at room temperature for 45 min. The slides were washed in TBS and incubated with diaminobenzidine tetrahydrochloride as the substrate, and counterstained with hematoxylin. A negative control without primary antibody was included in the experiment to verify the antibody specificity. Measurement of myocardial immunoreactivity for iNOS was performed in 100 randomly selected fields in heart sections in 200-fold magnification by light microscopy.
RNA extraction
Heart tissues were preserved by immersion in RNAlater (Ambion Inc, Austin, TX, USA) immediately after sampling. The extraction of total RNA was performed after homogenization using Ultra TurraxT8 (IKA Labortechnik, Staufen, Germany) in TRIzol reagent (Invitrogen Corporation, Carlsbad, CA, USA) in accordance with the standard protocol. Synthesis of cDNA was performed by reverse transcription using total RNA (2 μg) as a template (Super Script II; Invitrogen Corporation).
Gene expression analysis by realtime reverse transcription-polymerase chain reaction
Gene expression analysis was performed by real-time reverse transcription-polymerase chain reaction (RT-PCR) (Smart cycler; Cepheid, Sunnyvale, CA, USA) using cDNA synthesized from the DCM specimens. Realtime RT-PCR by monitoring with a TaqMan probe (TaqMan Gene expression assays; Applied Biosystems, Foster City, CA, USA) was performed in accordance with the following protocol: 600 s at 95°C, followed by thermal cycles of 15 s at 95°C, and 60 s at 60°C for extension. Relative standard curves representing several 10-fold dilutions (1:10:100:1000:10,000:100,000) of cDNA from DCM tissue samples (six samples/group) were used for linear regression analysis of other samples. Results were normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA as an internal control and are thus shown as relative mRNA levels.
Statistical analysis
All values are expressed as means ± SE. Statistical analysis of differences between the groups was performed by one-way analysis of variance, followed by Tukey's or Bonferroni's test and two-tailed t-test when appropriate. A value of P < 0.05 was considered statistically significant.
Results
Effects of olmesartan on myocardial functions
Although heart rate was not different among the three groups of rats, central venous pressure (CVP) and LVEDP were significantly higher and mean blood pressure (MBP), LVP and ±dP/dt were significantly lower in group V than in group N, indicating systolic and diastolic dysfunction in vehicle-treated rats (Table 1). CVP and LVEDP were significantly decreased in the Olm-10 treatment group compared with those in group V. Myocardial contractility parameters ±dP/dt were also improved in DCM rats treated with olmesartan. However, MBP and LVP were slightly improved by the treatment. Echocardiographic analysis of group V rats showed evidence of LV remodeling, with increased LVDd and LVDs (P < 0.01) and reduced FS and ejection fraction (EF) (P < 0.01), indicating impaired systolic function compared with that in group N rats (Table 1). Treatment with olmesartan significantly decreased LVDs and increased FS and EF compared with those in group V (Table 1).
Changes in hemodynamic, echocardiographic and histopathological parameters after four weeks of treatment with olmesartan in rats with DCM after EAM
N, no. of rats; BW, body weight; HW, heart weight; HW/BW, ratio of heart weight to body weight; CVP, central venous pressure; MBP, mean blood pressure; LVP, left ventricular pressure; LVEDP, left ventricular end-diastolic pressure; ±dP/dt, rate of intraventricular pressure rise and decline; HR, heart rate; LVDd, left ventricular dimension in diastole; LVDs, left ventricular dimension in systole; FS, fractional shortening; EF, ejection fraction; group N, aged-matched untreated rats; group V, rats with DCM treated with vehicle; group Olm-10, rats with DCM treated with olmesartan 10 mg/(kg/d)
Results are presented as the mean ± S.E
*P < 0.05 and **P < 0.01 versus group N; # P < 0.05 and ## P < 0.01 versus group V
Effects of olmesartan on Ang-II
The tissue Ang-II concentrations in group V did not differ from those in group N; however, plasma Ang-II concentrations were significantly elevated in group V in comparison to group N (Figure 1a). Treatment with olmesartan further significantly elevated the plasma concentration of Ang-II in comparison with that in group V (Figure 1a). In contrast, Ang-II level in the tissue was decreased in the olmesartan-treated group in comparison with that in group V (Figure 1b).
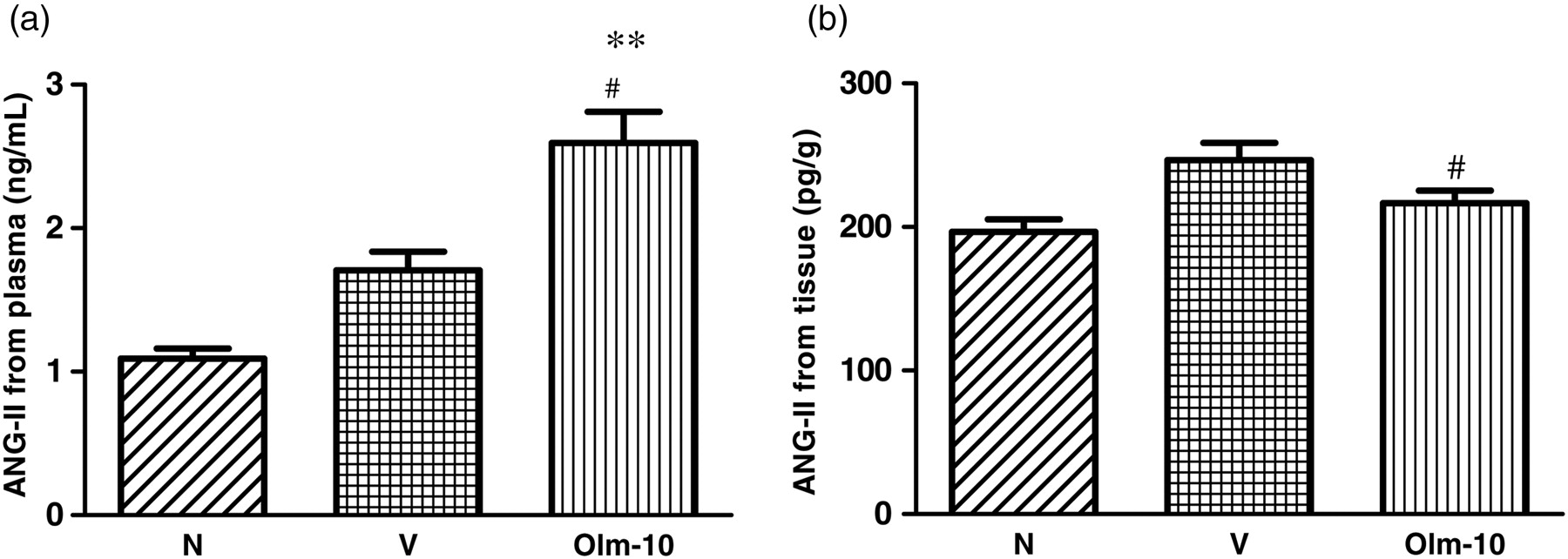
Effects of olmesartan on neurohumoral parameters in rats with DCM induced by autoimmune myocarditis. (a) Plasma Ang-II and (b) tissue Ang-II concentration. Group N, age-matched untreated rats; group V, DCM rats administered with vehicle; group Olm-10, DCM rats treated with olmesartan (10 mg/kg/d). The values are mean ± SEM. **P < 0.01 versus group N; # P < 0.05 and ## P < 0.01 versus group V. DCM, dilated cardiomyopathy; Ang, angiotensin; Olm-10, olmesartan-10
Effects of olmesartan on histopathology
HW and HW/BW were significantly larger in group V than in group N rats (Table 1). Olmesartan significantly reduced HW and HW/BW, when compared with those in group V. The hearts from group V rats showed massive fibrosis compared with those from group N rats (Figures 2a and a1). The percentage area of fibrosis was significantly lower in the olmesartan-treated rats than in vehicle-treated rats (Figures 2a and a1). The myocyte size in group V was significantly larger than that in group N (Figures 2b and b1). Olmesartan treatment significantly reduced the myocyte size compared with that in group V (Figures 2b and b1). Focal accumulations of inflammatory infiltrating cells were detected in the hearts of group V rats (Figure 2c). However, the proportion of cell accumulation was less in the olmesartan-treated rats (Figure 2c).
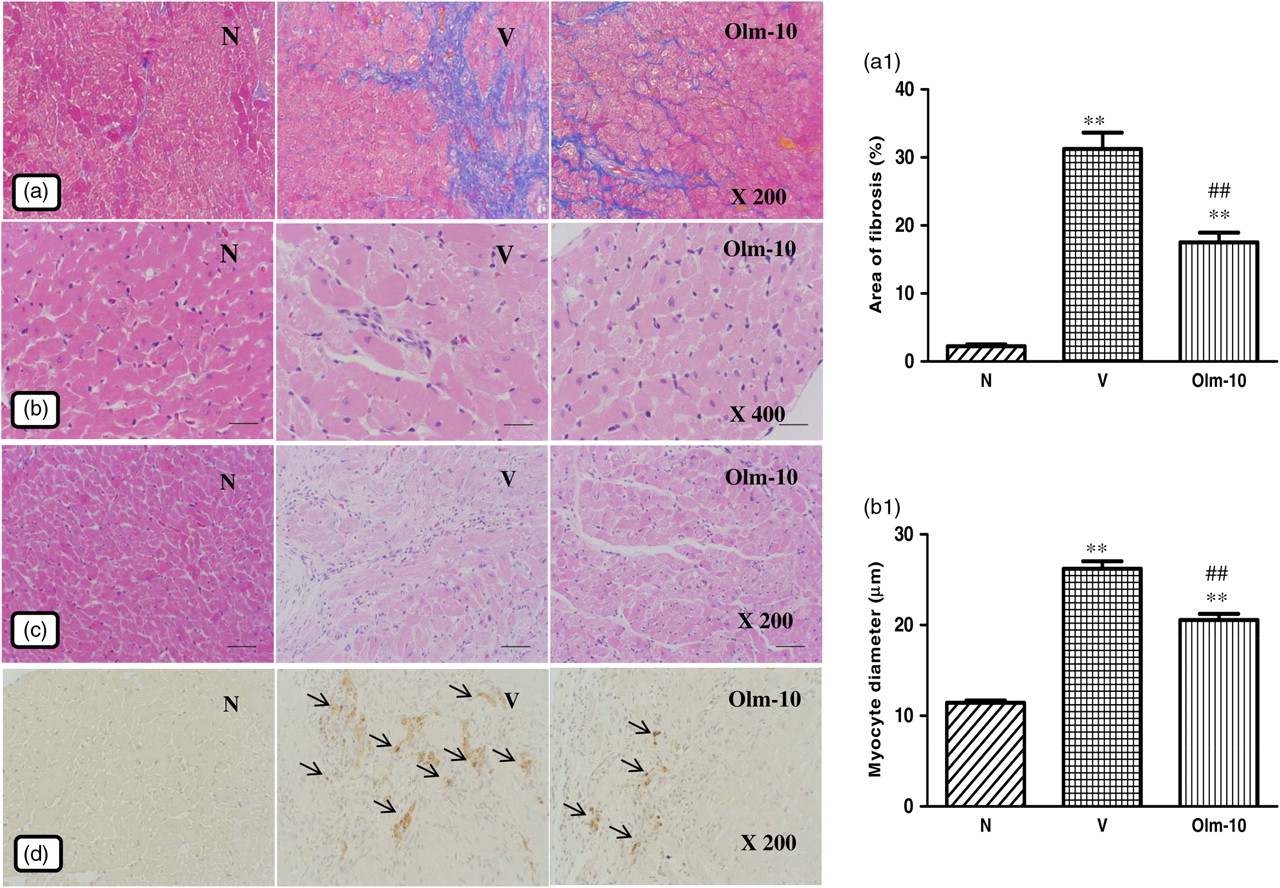
Effects of olmesartan on myocardial remodeling in rats with DCM induced by autoimmune myocarditis. (a) Azan–Mallory staining for fibrosis of the cross-sectional tissue slices of hearts. Fibrosis is indicated by the blue area contrasting to the red myocardium (×200). (b) Hematoxylin and eosin staining of the cross-sectional tissue slices of hearts depicting cardiomyocyte hypertrophy (×400). (c) Extensive inflammatory cell infiltration was observed in the hearts of myosin-immunized rats (hematoxylin–eosin staining; ×100) and (d) immunohistochemistry of iNOS (counterstained with hematoxylin; ×200). Bar graph shows quantitative analysis of fibrosis (a1) and myocyte diameter (b1) in groups N, V and Olm-10. Group N, age-matched untreated rats; group V, DCM rats administered with vehicle; group Olm-10, DCM rats treated with olmesartan (10 mg/kg/d). The values are mean ± SEM. *P < 0.05 and **P < 0.01 versus group N; ## P < 0.01 versus group V. DCM, dilated cardiomyopathy; iNOS, inducible nitric oxide synthase; Olm-10, olmesartan-10
Immunohistochemistry
Immunohistochemical analysis of rat hearts in group V showed enhanced expression of iNOS compared with that in normal rat hearts (Figure 2d). Myocardial immunoreactivity for iNOS was significantly decreased in the olmesartan-treated rats compared with that in the vehicle-treated rats (Figure 2d).
Effects of olmesartan on marker molecules of cardiac remodeling
RT-PCR analysis showed that collagen-I and -III, TGF-β1 and atrial natriuretic peptide (ANP) mRNA expression levels were significantly increased in vehicle-treated rats compared with those in group N rats (Figures 3a–d). In contrast, treatment with olmesartan significantly decreased the myocardial mRNA expressions of collagen-I and -III, TGF-β1 and ANP compared with those of group V rats (Figures 3a–d).
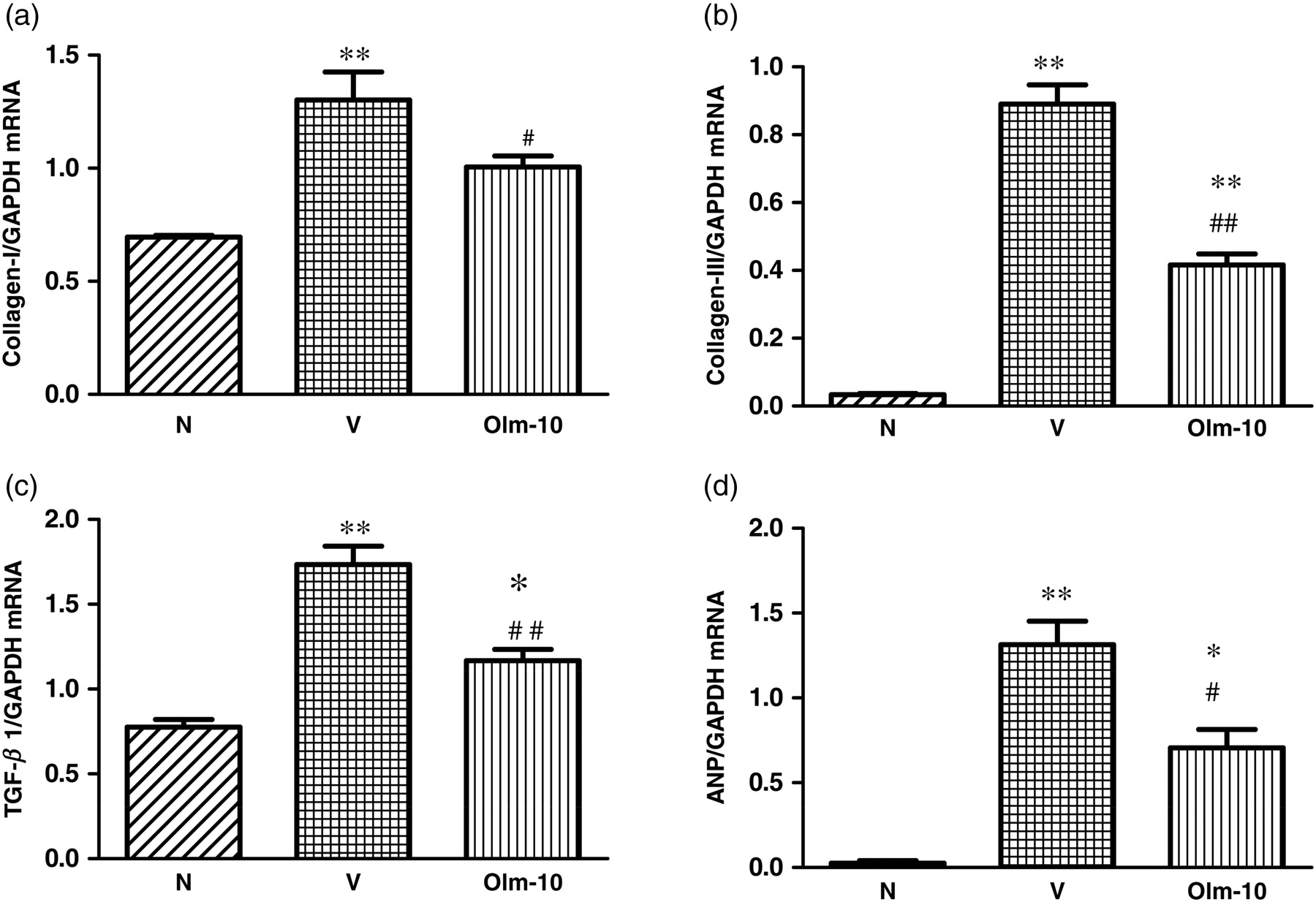
Myocardial messenger RNA expression levels of (a) collagen-I, (b) collagen-III, (c) TGF-β1 and (d) ANP in rats with DCM were determined by quantitative RT-PCR. The expression level of each sample was expressed relative to the expression level of GAPDH gene. Data are mean ± SEM of 4–6 rats. Group N, age-matched untreated rats; group V, DCM rats administered with vehicle; group Olm-10, DCM rats treated with olmesartan (10 mg/kg/d). The values are mean ± SEM. *P < 0.05 and **P < 0.01 versus group N; # P < 0.05 and ## P < 0.01 versus group V. DCM, dilated cardiomyopathy; TGF-β1, transforming growth factor beta1; ANP, atrial natriuretic peptide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Olm-10, olmesartan-10
Effects of olmesartan on inflammatory cytokines and matrix metalloproteinases
RT-PCR data showed that IL-6, IL-1β and monocyte chemoattractant protein-1 (MCP-1) mRNA expression levels were significantly increased in vehicle-treated rats compared with those in group N rats (Figures 4a–c). In contrast, treatment with olmesartan significantly decreased the myocardial mRNA expressions of IL-6, IL-1β and MCP-1 compared with those of group V rats (Figures 4a–c). Moreover, mRNA expressions of matrix metalloproteinase (MMP)-2 and -9 were also significantly increased in vehicle-treated rats compared with those of group N rats (Figures 5a and b). However, treatment with olmesartan significantly reduced the myocardial mRNA expressions of MMP-2 and -9 compared with those of group V rats (Figures 5a and b).
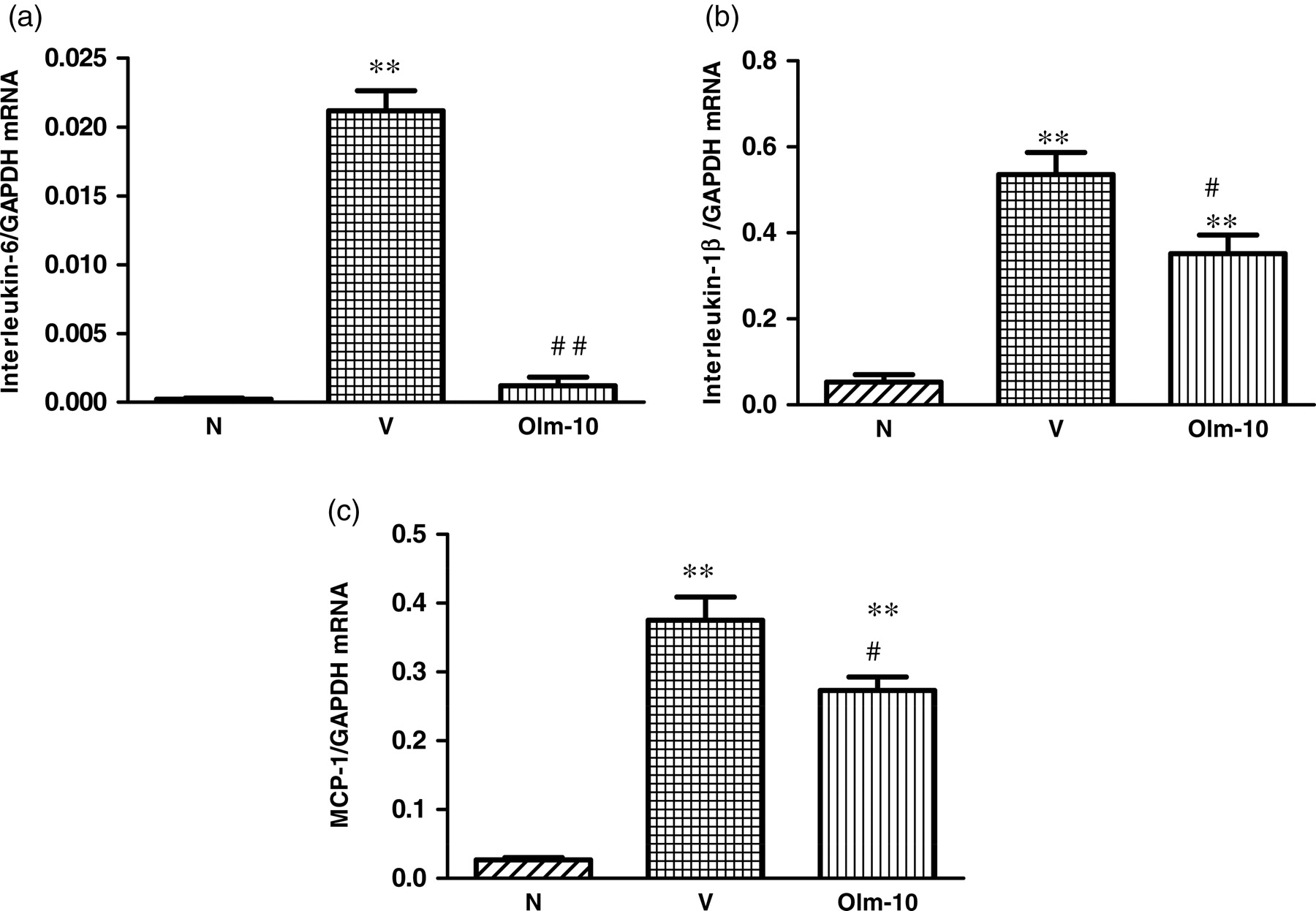
Myocardial messenger RNA expression levels of (a) IL-6, (b) IL-1β, and (c) MCP-1 in rats with DCM were determined by quantitative RT-PCR. The expression level of each sample was expressed relative to the expression level of GAPDH gene. Data are mean ± SEM of 4–6 rats. Group N, age-matched untreated rats; group V, DCM rats administered with vehicle; group Olm-10, DCM rats treated with olmesartan (10 mg/kg/d). The values are mean ± SEM. **P < 0.01 versus group N; # P < 0.05 and ## P < 0.01 versus group V. DCM, dilated cardiomyopathy; IL, interleukin; MCP-1, monocyte chemoattractant protein-1
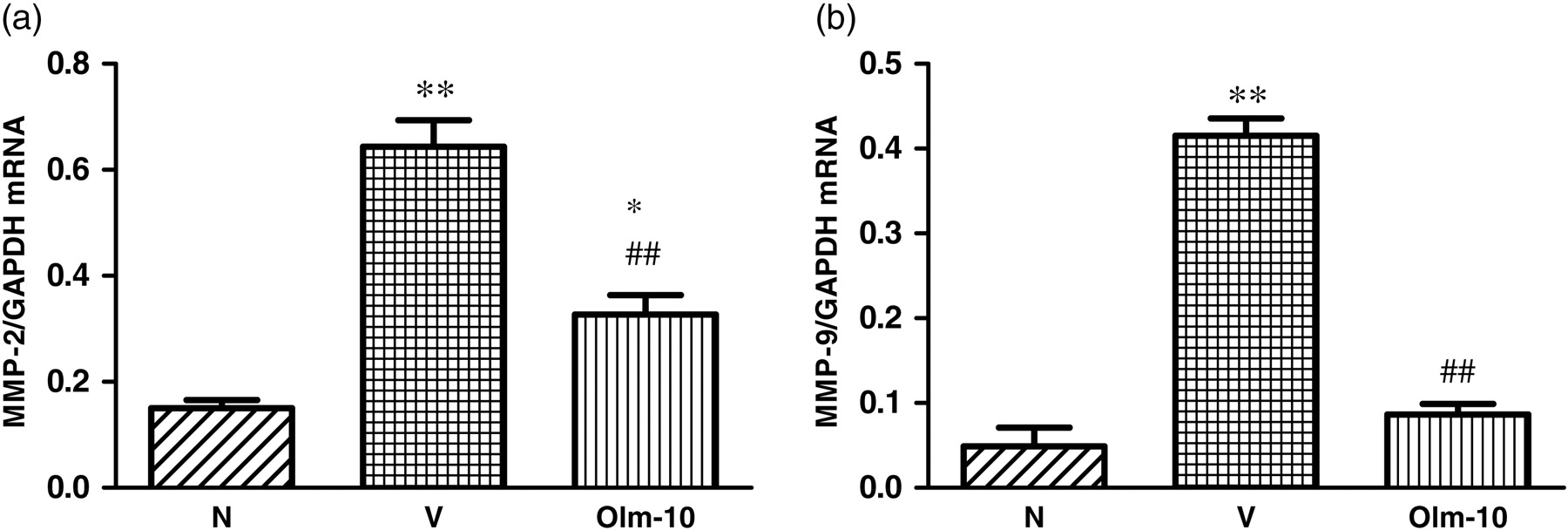
Myocardial messenger RNA expression levels of (a) MMP-2 and (b) MMP-9 in rats with DCM were determined by quantitative RT-PCR. The expression level of each sample was expressed relative to the expression level of GAPDH gene. Data are mean ± SEM of 4–6 rats. Group N, age-matched untreated rats; group V, DCM rats administered with vehicle; group Olm-10, DCM rats treated with olmesartan (10 mg/kg/d), respectively. The values are mean ± SEM. *P < 0.05 and **P < 0.01 versus group N; ## P < 0.01 versus group V. MMP, matrix metalloproteinase; DCM, dilated cardiomyopathy; RT-PCR, reverse transcription-polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Olm-10, olmesartan-10
Discussion
The results of this study demonstrate that the treatment with oral olmesartan improved both systolic (+dP/dt, % EF and % FS) and diastolic (−dP/dt and LVEDP) functions, increased neurohormonal parameter, such as plasma Ang-II, and ameliorated myocardial remodeling (fibrosis and hypertrophy) and its marker molecules.
Myocardial fibrosis probably plays an important role in both diastolic and systolic dysfunction, 23 and has adverse clinical consequences that result in increased mortality caused by progressive heart failure. It has been proposed that the increase in myocardial fibrosis during heart failure is due to both increased collagen synthesis by fibroblasts and unchanged or decreased fibrillar collagen degradation. 24 Myocardial fibrosis, the hallmark of DCM, is observed in DCM hearts as indicated by Azan–Mallory staining and increased concentrations of its marker molecules (TGF-β1, collagen-I and -III) (Figures 2 and 3a–c). It has been reported that AT1R blockade reduces myocardial hypertrophy, decreases myocardial fibrosis and attenuates cardiac remodeling, which support the hypothesis that Ang-II plays a role in fibrous tissue formation by promoting TGF-β1 synthesis via AT1R. 16,17,25–27 Consistent with previous studies, 16,17,25–27 we could also observe increased expressions of TGF-β1 and collagen-I and -III mRNA in group V, and this increased mRNA was suppressed by olmesartan treatment (Figures 3a–c). Taken together, these findings suggest that AT1R has an important role in cardiac remodeling via expression of TGF-β1 and collagen-I and -III, and that olmesartan may contribute to the inhibition of cardiac remodeling. However, further studies are required to investigate the precise mechanism of the antifibrotic effects of olmesartan.
Myocardial ECM remodeling plays an important role in the development of LV hypertrophy and heart failure. The heart contains many MMPs, including MMP-2, MMP-3 and MMP-9, which can degrade ECM proteins with differing degrees of specificity. 28 It has been reported that local activation of Ang-II, a critical effector in the RAS in hypertrophic LV, triggers ECM degradation, resulting in LV remodeling. 26,29 Treatment with olmesartan preserves LV shape and function, as well as ECM density, and decreases oxidative stress-mediated protein degeneration. 30 This evidence supports the assertion that AT1R blockade is a potential strategy to normalize the MMP/tissue inhibitor of metalloproteinase-1 balance. 31 Interestingly, we could observe an increase in the myocardial mRNA levels of MMP-2 and -9 in the DCM rats (Figure 5). In agreement with the previous study, 30 olmesartan treatment significantly decreased myocardial mRNA levels of MMP-2 and -9, resulting in improvement in myocardial function, which in turn supports the notion that the AT1R is involved in ECM remodeling.
It is believed that the administration of AT1R blockers increases the Ang-II level because of a lack of negative feedback of renin. Studies have shown that most AT1R blockers increase renin activity and/or plasma Ang-II level. 32,33 In contrast, Ichikawa and Takayama 34 reported that long-term treatment of hypertensive patients with olmesartan resulted in a reduction in the plasma Ang-II level, despite the fact that several types of AT1R blockers have been shown to increase plasma Ang-II concentrations in hypertensive patients, although the mechanism of this effect was not clarified. 35 Interestingly, we could observe elevated Ang-II in the olmesartan-treated rats compared with that in the DCM rats. These results support the hypothesis that the increased Ang-II plasma levels by AT1R blockade are indicative of a high potential for Ang AT2R stimulation, which results in an antifibrotic effect.
Recent investigations have shown that AT1R blockade decreased cardiac tissue Ang-II levels and increased circulating Ang-II levels in rats with myocardial infarction, supporting the role of cardiac-derived Ang for the development of ventricular hypertrophy. 36 In this study, tissue Ang-II levels also decreased during long-term treatment with olmesartan, which may support the role of cardiac-derived Ang-II in the development of cardiac remodeling. However, since the effect of AT2R stimulation on various tissues during treatment with AT1R blockers is still unknown, further investigation is necessary to determine the nature of these events.
In patients with heart failure, the plasma level of ANP is documented to be elevated, the level being related to the severity of heart failure. 37 Interestingly, we could observe that vehicle-treated rats had developed cardiac hypertrophy and LV dilation, evidenced by an increase in mRNA levels of ANP (Figure 3d), myocyte size, HW, HW/BW, LV diastolic and systolic dimension, and a decrease in FS (Figure 2 and Table 1). A significant reduction in these parameters and increased FS were seen in the olmesartan-treated rats. Taken together, these results indicate that olmesartan improved LV function and ameliorated the progression of cardiac remodeling.
Several clinical studies have described the participation of proinflammatory cytokines in the pathogenesis of cardiac diseases. 5,13,38,39 The levels of circulating proinflammatory cytokines such as tumor necrosis factor-α and IL-1 and -6 are elevated in patients with myocarditis. 38,39 It has been reported that Ang-II induces inflammation through the production of reactive oxygen species (ROS), adhesion molecules and inflammatory cytokines such as MCP-1. 40 AT1R antagonists are reported to suppress cytokine production and the transcription of cytokine genes in vitro and in vivo, and significantly decrease MCP-1 and iNOS expression, which further supports the notion that Ang-II induces inflammation through the production of ROS and inflammatory cytokines. 14,15,38–41 Interestingly, we could observe an increase in the myocardial mRNA levels of IL-6, IL-1β and MCP-1, and increased inflammatory cell infiltration and iNOS expression in rats with DCM; these changes were significantly decreased by olmesartan treatment (Figures 2 and 4). These results suggest that the beneficial effects of olmesartan in EAM may be partly due to the suppression of inflammatory events in the myocardium. Furthermore, olmesartan treatment might be a promising new therapy for chronic myocarditis, particularly for LV remodeling where ongoing autoimmune processes may play a role in the disease development.
Human DCM is thought to have a variety of causes. Therefore, the clinical courses and pathological findings in this cardiomyopathy are not uniform. Cardiac myosin-induced EAM, which is not exclusively related to viral infection, develops clinicopathologically to resemble DCM in the chronic phase. 2 Thus, the present results provide some insight into the effectiveness of olmesartan treatment against DCM after EAM.
In summary, this study demonstrated that olmesartan treatment ameliorated the progression of rat EAM. These results may have particular clinical relevance because olmesartan is safely used in the clinical field. The olmesartan dose used in the study is clinically relevant. Thus, olmesartan may exert beneficial effects on the clinical course of patients with heart failure. Further study is needed, but this study suggests the beneficial effect of olmesartan for patients with DCM.
Footnotes
Acknowledgements
This research was supported by a Yujin Memorial Grant, Ministry of Education, Culture, Sports, Science and Technology, Japan, and by a grant from the Promotion and Mutual Aid Corporation for Private Schools, Japan. We thank Wawaimuli Arozal, Flori Ratna Sari, Hiroko Shimazaki, Sayaka Egawa, Fuji Tomohiko, Kana Kawadura, Yoshiyasu Kobayashi and Yuhki for their assistance in this research work. We also express our sincere gratitude to Dr Masaki Nagata (Division of Oral and Maxillofacial Surgery, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan) for help with the RT-PCR analysis in this study.