
Abstract
The aim of this study was to investigate the possible proinflammatory signaling pathways involved in statin inhibition of glucose-induced plasminogen activator inhibitor-1 (PAI-1) expression in cardiac microvascular endothelial cells (CMECs). Primary rat CMECs were grown in the presence of 5.7 or 23 mmol/L glucose. PAI-1 mRNA and protein expression levels were measured by realtime polymerase chain reaction, Western blotting and enzyme-linked immunosorbent assay, respectively. A pull-down assay was performed to determine RhoA activity. IkBa protein expression was measured by Western blotting, nuclear factor (NF)-kB activation was detected by electrophoretic mobility shift assay and its transcription activity was determined by a dual luciferase reporter gene assay. PAI-1 mRNA and protein expression levels were both increased with high glucose concentrations, but they were significantly suppressed by simvastatin and atorvastatin treatment (P, 0.01) and the effects were reversed by mevalonate (100 mmol/L) and geranylgeranyl pyrophosphate (10 μmol/L) but not farnesyl pyrophosphate (10 μmol/L). Such effects were similar to those of a RhoA inhibitor, C3 exoenzyme (5 μg/mL), inhibitors of RhoA kinase (ROCK), Y-27632 (10 μmol/L) and hydroxyfasudil (10 μmol/L) and an NF-kB inhibitor, BAY 11-7082 (5mmol/L). High glucose-induced RhoA and NF-kB activations in CMECs were both significantly inhibited by statins (P, 0.01). Simvastatin and atorvastatin equally suppress high glucose-induced PAI-1 expression. These effects of statins may occur partly by regulating the RhoA/ROCK-NF-kB pathway. The multifunctional roles of statins may be particularly beneficial for patients with metabolic syndrome.
Introduction
The dynamic equilibrium of coagulation and fibrinolysis systems plays a pivotal role in maintaining hemostasis and preventing pathological thrombogenicity. Plasminogen activator inhibitor-1 (PAI-1) is the major physiological modulator of fibrinolysis and is produced mainly by endo-thelial cells. Endothelial dysfunction has been known to be a critical factor of the onset of diabetic vascular complications. 1 Impaired endothelial functions may lead to disturbances in the balance of coagulation and fibrinolysis in the early stages of poor glucose metabolism, which contributes to the development and progression of local inflammation and eventually atherosclerosis. 2 Numerous clinical investigations have demonstrated that PAI-1 is one of the risk factors of cardiovascular events such as acute coronary syndrome. Elevated PAI-1 levels are also associated with the pathogenesis of stroke, diabetes and metabolic syndrome. 3 In addition, plasma PAI-1 is a major independent predictor of coronary microvascular dysfunction and may be involved in vascular tone regulation. 4 Furthermore, impaired cardiac microvascular endothelial cell (CMEC) function with abnormal PAI-1 levels may lead to aggravation of myocardial ischemia in coronary artery diseases (CAD). 5
In addition to their lipid-lowering effects as HMG-CoA reductase inhibitors, statins have been suggested to act as anti-inflammation agents and hemostasis regulators. In early glucose metabolism-impaired patients, simvastatin was shown to reduce fibrinogen, PAI-1 levels and factor VII activity, whereas it prolonged the prothrombin and partial thromboplastin time in a lipid- and glucose-independent manner. 2 Atorvastatin appears to have effects on coagulation activation, fibrinolysis and inflammation in patients with CAD by lowering plasma levels of thrombin–antithrombin complexes, PAI-1 activity and inflammatory factors.6,7 However, the mechanism of statin regulation on coagulation and fibrinolysis remains unclear.
The small GTPase RhoA belongs to the Rho subfamily, which also includes RhoB and RhoC. RhoA switches between the inactive GDP-bound and the active GTP-bound conformations, which are regulated by guanosine nucleotide exchange factors (GEFs). Rho-kinases (ROCKs) are the first and the best-characterized RhoA effectors. ROCKs are activated by phosphorylation of several amino acid sites after receiving the Rho activation signals, and then they mediate a series of downstream phosphorylation/ dephosphorylation signal cascades to regulate a variety of pathophysiological conditions such as inflammation, atherosclerosis, thrombus formation, vascular spasm and reperfusion injury.8–10 Statins have been suggested to inhibit the synthesis of geranylgeranyl pyrophosphate (GGPP) and farnesylpyrophosphate (FPP), which are downstream of mevalonate (MVA). Small GTP-binding proteins are modificated by GGPP and FPP; 11 therefore, the iso-prenylation of small GTP-binding proteins is speculated to be influenced by statins. To date, whether statins suppress or increase the active form of RhoA or have no effects at all remains very controversial.11–15
Nuclear factor (NF)-κB plays an important role in inflammation, cell proliferation, atherosclerosis and angio-genesis. 16 Activated NF-κB is associated with endothelial dysfunction, whereas statins are partially anti-inflammatory through inhibition of NF-κB. 17 Whether NF-κB plays a role in the signaling pathway by which statins suppress the production of PAI-1 in endothelial cells is unclear. The aims of this study were to investigate the involvement of RhoA/ ROCK and NF-κB pathways in the suppressive effect of statins on high glucose-induced PAI-1 expression in a primary culture of rat CMECs, and to clarify the possible relationship between the RhoA and the NF-κB signaling pathways.
Materials and methods
Animals
Adult male Sprague-Dawley rats (weighing 150–200 g) were obtained from Nantong University, China. The study was approved by the local animal ethics review board of Nantong University.
Materials
Simvastatin, atorvastatin prodrugs, MVA and its metabolic product GGPP and FPP were obtained from Sigma Aldrich (St Louis, MO, USA). ROCK inhibitors, Y-27632 and hydroxyfasudil, and Rho inhibitor C3 exoenzyme were purchased from Calbiochem (Darmstadt, Germany). The NF-κB inhibitor BAY 11-7082 was obtained from Beyotime (Nantong, China).
Isolation and characterization of CMECs
CMECs were isolated according to the procedure of Nishida et al. 18 Briefly, under anesthesia (by 1% sodium pentobarbital, 30 mg/kg), rat hearts were removed and washed with ice cold Hanks’ balanced salt solution (without Ca2+ and Mg2+). The outer one-fourth of the left ventricle free wall and septum was dissected to remove epicardial arteries and larger penetrating vessels. The remaining tissue was minced in 0.2% collagenase II (Sigma, St Louis, MO, USA) and incubated for 30 min at 37°C in a shaking bath. Trypsin (0.02%, Sigma) was added, and the tissue was sheared 10 times over a period of 30 min. Dissociated cells were filtered through a 100 μm mesh filter, washed with Hanks’ balanced salt solution and centrifuged at 100g for five minutes. Cells were re-suspended in Dulbecco's modified Eagle's medium (DMEM). After a four-hour period, attached cells were washed with DMEM to allow differential adhesion. The CMECs were plated on gelatin-coated culture dishes at a density of approximately 2.5 × 10 3 cells/cm2 . CMECs were cultured in DMEM containing 5.7 mmol/L glucose supplemented with 10% fetal bovine serum (Gibco, Paisley, Scotland, UK) and 1% (V/V) endothelial cell growth supplement (Sciencell, San Diego, CA, USA) in 95% O2/5% CO2 at 37°C. Relatively pure (>98%) CMECs were confirmed by their characteristic cobblestone morphology when they reached confluence and immuno-cytochemical staining for von Willebrand factor and CD31.
Cell treatments
Low glucose DMEM contained 5.5 mmol/L glucose (1000 mg/L) and high glucose DMEM contained 25 mmol/L glucose (4500 mg/L). By adding serum containing 1.26 g/L D-glucose, the final concentration of glucose in DMEM reached 5.7 and 23 mmol/L, respectively. According to the method of Sadeghi et al.,19 simvastatin or atorvastatin was dissolved in DMSO or 100% ethanol, respectively, at a concentration of 10 mmol/L after being activated by NaOH at 50°C for two hours followed by adjusting the pH to 7.0 with HCl. The concentration of vehicle did not exceed 0.1% (V/V). The stock solution was kept at -70°C for extended storage or at 4°C for immediate use. Before treatments, the CMECs were switched to serum-free medium for 12 h. For statin/isoprenoid or RhoA/NF-κB inhibitor treatment, CMECs were preincubated in low glucose medium for 30 min with statin/isoprenoids or each inhibitor; then, the medium was changed to high glucose medium with the indicated doses of statin/isoprenoid/ inhibitor for further incubation.
Determination of cell viability
The viability of CMECs for further experiments within 2–4 passages was examined by trypan blue exclusion. In order to determine possible cytotoxic effects of the statins, we measured lactate dehydrogenase (LDH) leakage of conditioned cells according to the manufacturer's instructions (Jiancheng Bioengineering Institute, Nanjing, China).
Enzyme-linked immunosorbent assay for PAI-1 measurements
PAI-1 levels in conditioned medium from CMECs were measured with an enzyme-linked immunosorbent assay (ELISA) obtained from Sun Biotech (Shanghai, China). The conditioned media samples were collected and centrifuged at 18,000g for five minutes. PAI-1 in the supernatant was measured according to the manufacturer's protocol. The quantities of PAI-1 in the test samples were determined using a standard curve generated with purified recombinant PAI-1 provided with the kit.
RNA isolation and quantitative reverse transcription polymerase chain reaction
We used two-step quantitative reverse transcription polymerase chain reaction (PCR) with SYBR Green I for gene expression analysis. Total cellular RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcribed using the PrimeScript RT reagent kit with gDNA Eraser (TaKaRa, Shiga, Japan). PCR amplification was performed using the SYBR Premix Ex Taq II kit (TaKaRa). We used the ΔΔCT method and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal standard to correct for potential variation in RNA loading, cDNA synthesis or efficiency of PCR amplification. The oli-gonucleotide sequences used for PAI-1 were as follows: forward primer, 5’ TCA TCA TCA ATG ACT GGG TGA AGA C 3'; reverse primer, 5’ TTC CAC TGG CCG TTG AAG TAG AG 3’. The primers used for GAPDH were as follows: forward primer, 5’ GGT GAA GGT CGG AGT CAA C 3'; reverse primer, 5’ CCA TGG GTG GAA TCA TAT TG 3’ . The reaction cycles for PCR were 95°C for 10 s, 95°C for five seconds and 60°C for 30 s for 40 cycles.
Determination of RhoA activation
RhoA activity was measured using a pull-down assay with the Rho binding domain of the Rho effector protein Rhotekin (Rhotekin RBD), according to the manufacturer's protocol (Cytoskeleton, Denver, CO, USA). Briefly, cells were rinsed three times with ice-cold phosphate-buffered saline (PBS), and cell lysis buffer (50 mmol/L Tris [pH 7.5], 10 mmol/L MgCl2, 0.5 mol/L NaCl and 2% Igepal) was added. After incubating on ice for 10 min, the cells were collected and immediately clarified by centrifugation at 10,000 rpm and 4°C for two minutes. Protein (300–800 μg) was incubated for one hour at 4°C on a rotator with 50 μg of Rhotekin-RBD beads to precipitate GTP-bound Rho. The beads were washed once with wash buffer (25 mmol/L Tris [pH 7.5], 30 mmol/L MgCl2 and 40 mmol/L NaCl) and boiled for six minutes in loading buffer. Total cell extracts and precipitates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electro-phoresis (SDS–PAGE) and Western blot analysis using a monoclonal antibody against RhoA at a dilution of 1:500.
Western blot analysis
Cells were rinsed three times with PBS and lysised with Radio Immunoprecipitation Assay (RIPA) buffer supplemented with phenylmethylsulfonyl fluoride (1% V/V). Cells were then incubated for 20 min on ice, scraped and centrifuged. Supernatant protein concentrations were determined by BCA Protein Assay (Beyotime). Equal amounts of protein (20 μg) were solubilized in loading buffer, boiled for six minutes, separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA, USA). After incubating with blocking solution at room temperature for 30 min, the membranes were probed at 4°C overnight with primary antibodies for RhoA (1:500, Cytoskeleton), IΚBα or PAI-1 (1:500, Santa Cruz Biotechnology, Inc.) and β-actin (1:1000, Beyotime) in Tris-buffered saline-Tween. The membranes were incubated with the appropriate secondary antibodies (1:5000) for one hour at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescence reaction (ECL, Millipore Corp.).
Electrophoretic mobility shift assay
The levels of NF-κB proteins in nuclear extracts from the cells were analyzed by electrophoretic mobility shift assay (EMSA). CMECs were preincubated without or with simvastatin (10 μmol/L), atorvastatin (10 μmol/L), C3 exo-enzyme (5 μg/mL), Y-27632 (10 μmol/L), hydroxyfasudil (10 μmol/L) or BAY 11-7082 (5 μmol/L) for 30 min, and treatment continued with high glucose stimulation for six hours. Nuclear proteins from these cells were isolated and subjected to EMSA using digoxigenin (DIG)-labeled NF-κB doublestrand oligonucleotide (5’ AGT TGA GGG GAC TTT CCC AGG C 3’) for 30 min, subjected to gel electrophoresis and finally autoradiographed.
NF-κB reporter assay
CMECs were co-transfected with 0.5 μg of pNF-κB-Luc reporter plasmid (Stratagene, La Jolla, CA, USA), which contains the 5 × NF-κB binding site as an enhancer with the firefly luciferase reporter gene, along with 0.01 μg of Renilla luciferase (pRL-TK) using Fugene HD according to the manufacturer's procedure. Twenty-four hours after transfection, the cells were pretreated with or without statins or a specific inhibitor for 30 min and then stimulated with high glucose for 12 h. Luciferase and Renilla luciferase were measured using the Dual-Luciferase Reporter Assay Kit (Promega, Fitchburg, WI, USA). Results were presented as luciferase activity normalized to Renilla luciferase activity.
Statistical analysis
Data are expressed as means ± standard deviation (SD). All data were analyzed by two-way ANOVA or the Student's t-test as appropriate. A P value less than 0.05 was considered to be statistically significant.
Results
Effects of statins on cell viability
Preconfluent CMECs were treated with different concentrations of statins (0.1–30 μmol/L) for 30 min before high glucose (23 μmol/L) was added in the presence of the same concentration of statins. The cells were incubated for an additional 24 h. Conditioned media samples were assayed for LDH activity. There was no statistical difference between the LDH activities of cells exposed to 0.1–10 μmol/L simvastatin or atorvastatin compared with controls (high glucose without statin) (P > 0.05). However, when cells were treated with 30 μmol/L simvastatin or atorvastatin for 24 h, significant increases of LDH leakage were observed. The LDH activity with 30 μmol/L simvastatin treatment was 345.5 ±18.0 U/L versus that of the control was 274.7±5.9 U/L (P < 0.05); the LDH activity with 30 μmol/L atorvastatin treatment was 331.1 ± 8.3 U/L versus that of the control was 223.9 ± 24.7 U/L (P, 0.01), suggesting possible cytotoxicity due to high-dose statin treatment.
Trypan blue exclusion assays were performed at the end of most experiments. All groups demonstrated >95% viability, suggesting no differences between control and statin-treated groups.
PAI-1 mRNA and protein expressions induced by high glucose
High glucose (23 mmol/L) treatment induced a time-dependent increase of PAI-1 mRNA and protein expression levels from six hours post-treatment (P < 0.05), the mRNA expression levels peaked after 12 h and the protein expression levels peaked after 24–72 h (all P < 0.01) (Figures 1a and b). The PAI-1 mRNA and protein levels significantly increased at 24 h after high glucose stimulation; therefore, we chose 24 h as the time point to evaluate the effect of statins in the following experiments. Glucose-induced PAI-1 mRNA and protein expressions also demonstrated a dose-dependent increase with glucose concentrations ranging from 5.7 to 23 mmol/L for stimulation for 24 h (Figures 1c and d).
Plasminogen activator inhibitor-1 (PAI-1) 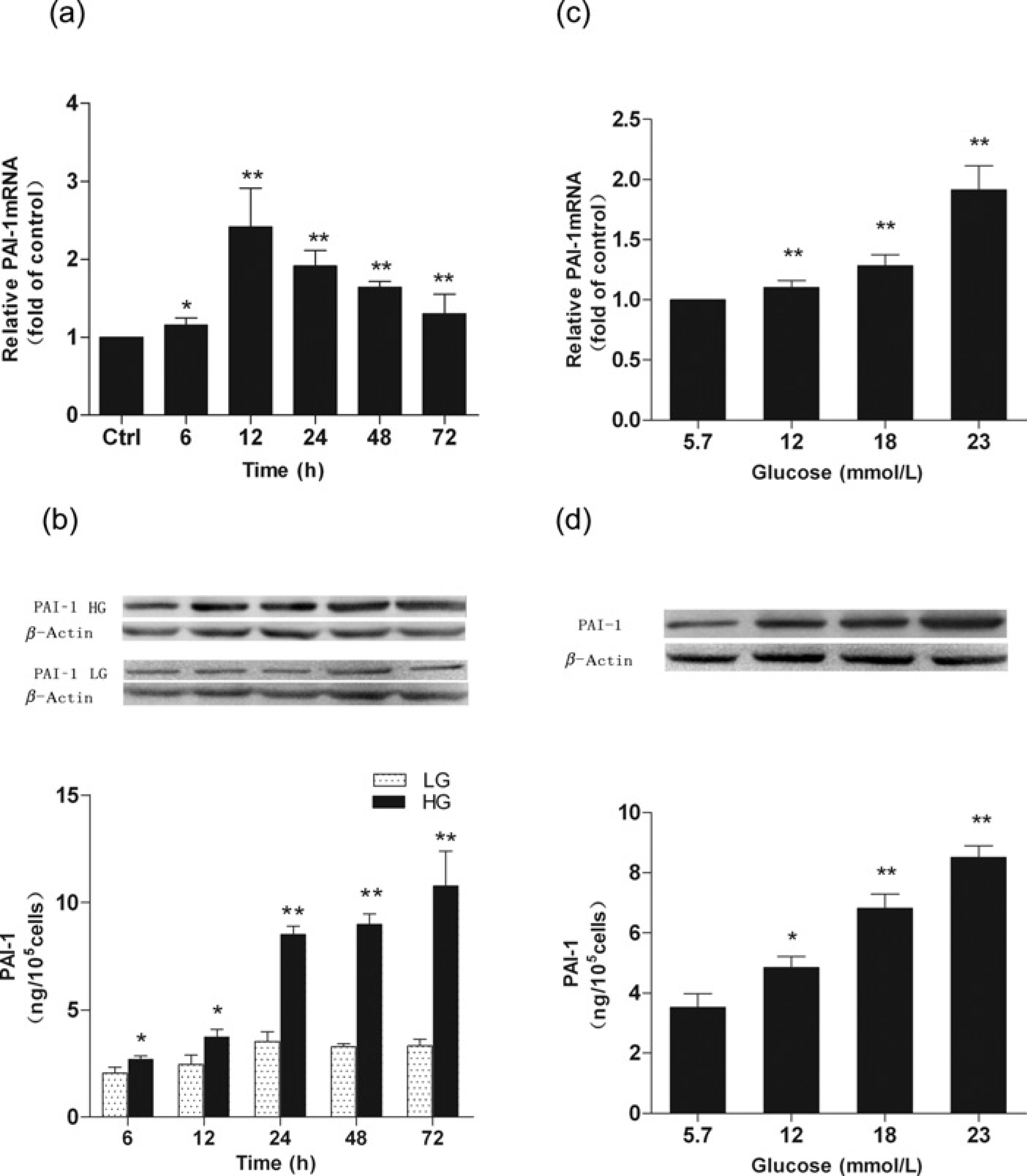
Effects of simvastatin and atorvastatin on high glucose-induced PAI-1 mRNA and protein expressions
Both statins markedly reduced PAI-1 mRNA expression at 0.1–10 μmol/L concentrations, compared with cells treated only with high glucose or statin vehicle (Figures 2a and b). The reduction effects were in a dose-dependent manner in that the 10 μmol/L statins caused high glucose-induced PAI-1 mRNA increases to be suppressed to levels comparable to those subjected to low glucose treatment. Simvastatin and atorvastatin also demonstrated a dose-dependent suppression of high glucose-induced PAI-1 protein expression increases (Figures 2c and d). Simvastatin and atorvastain at 1and 10 μmol/L caused significant suppression of PAI-1 protein expression (P < 0.01). Pretreatment of CMECs with GGPP (10 μmol/L) or MAV (100 μmol/L) but not FPP (10 μmol/L) reversed the inhibitory effect of simvastatin and atorvastatin on PAI-1 mRNA (Figures 2a and b) and protein expression (Figures 2c and d) induced by high glucose.
Effects of simvastatin or atorvastatin on plasminogen activator inhibitor-1 (PAI-1) mRNA and protein expressions. Cardiac microvascular endothelial cell were pretreated with 0.1 -10 μmol/L statins for 30 min before high glucose stimulation in the presence of statins for 24 h. (a,b) Both statins suppressed high glucose (23 mmol/L)-induced PAI-1 mRNA expression in a dose-dependent manner. (C,d) Both statins suppressed high glucose (23 mmol/L)-induced PAI-1 protein expression in a dose-dependent manner. Such effects can be reversed by geranylgeranyl pyrophosphate (10 μmol/L), mevalonate (100 μmol/L) but not farnesyl pyrophosphate (10 μmol/L). LG, low glucose (5.7 mmol/L); HG, high glucose (23 mmol/L). Data are expressed as means ± SD (n = 3). *P < 0.05, **P < 0.01 versus HG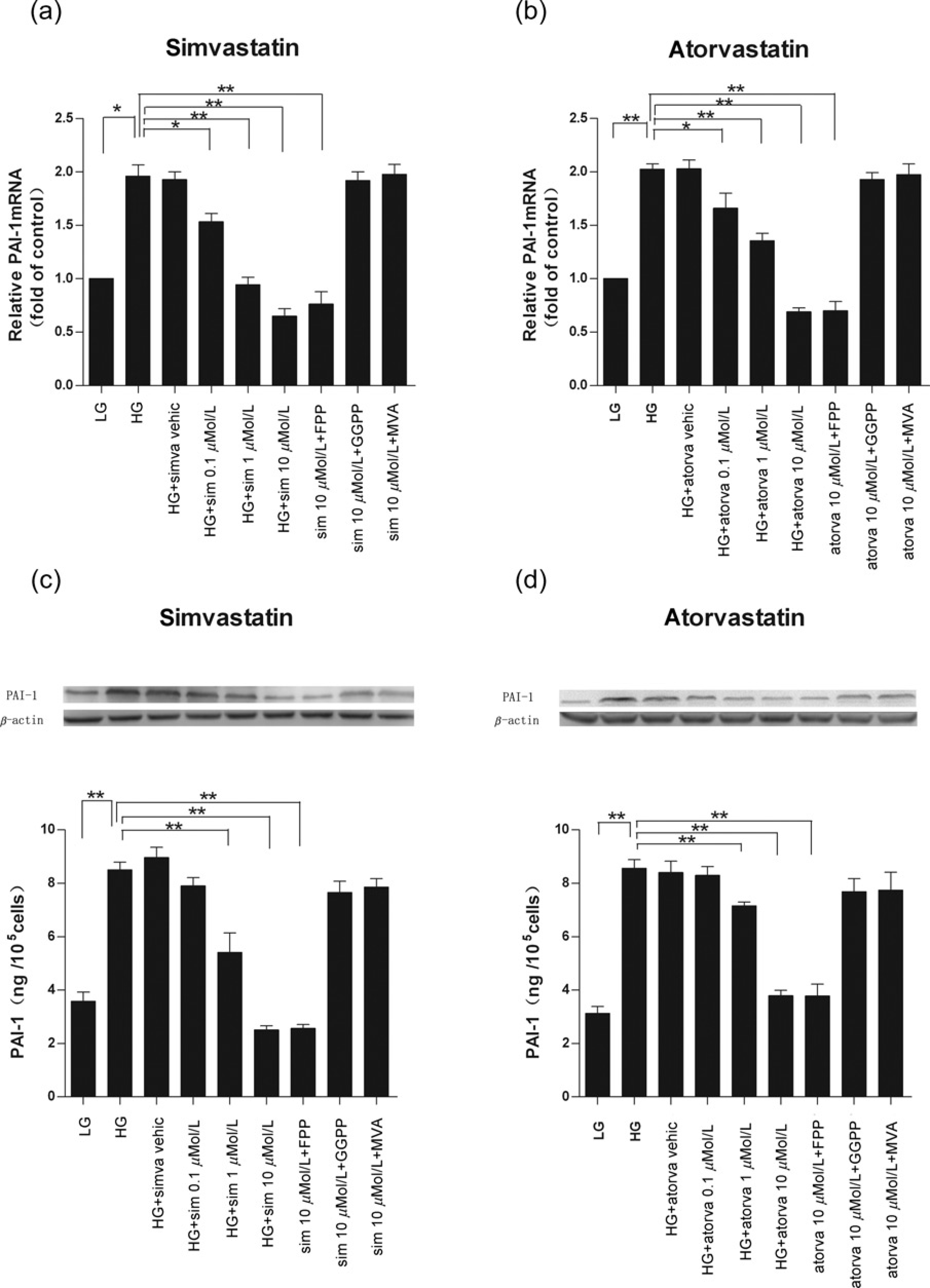
Effects of Rho/ROCK and NF-κB inhibitors on high glucose-induced PAI-1 mRNA and protein expressions
C3 transferase exoenzyme is an exotoxin produced by Clostridium botulinum that specifically inhibits Rho small GTP binding proteins (RhoA, RhoB and RhoC), Y-27632 is a non-selective inhibitor of ROCK1 and ROCK2, and hydroxyfasudil is a highly selective inhibitor of ROCK. Preincubation of CMECs with 5 μg/mL C3 exoenzyme, 10 μmol/L Y-27632 or 10 μmol/L hydroxyfasudil for 30 min and continued treatment with these inhibitors along with high glucose for 24 h significantly suppressed elevated PAI-1 mRNA and protein expression levels (all P < 0.01) (Figures 3a and b). Treatment with C3 exoenzyme, Y-27632 or hydroxyfasudil did not affect cell viability (data not shown).
Effects of inhibitors of Rho/ROCK or NF-κB on high glucose-induced plasminogen activator inhibitor-1 (PAI-1) mRNA and protein expressions. Cardiac microvascular endothelial cells were preincu-bated without or with C3 exoenzyme (5 μg/mL), Y-27632 (10 μmol/L), hydro-xyfasudil (10 μmol/L) or BAY 11-7082 (5 μmol/L) for 30 min, and treatment continued with high glucose stimulation for 24 h. (a) PAI-1 mRNA expression; (b) PAI-1 protein expression. LG, low glucose (5.7 mmol/L); HG, high glucose (23 mmol/L). Data are expressed as means ± SD (n = 3). **P < 0.01 versus HG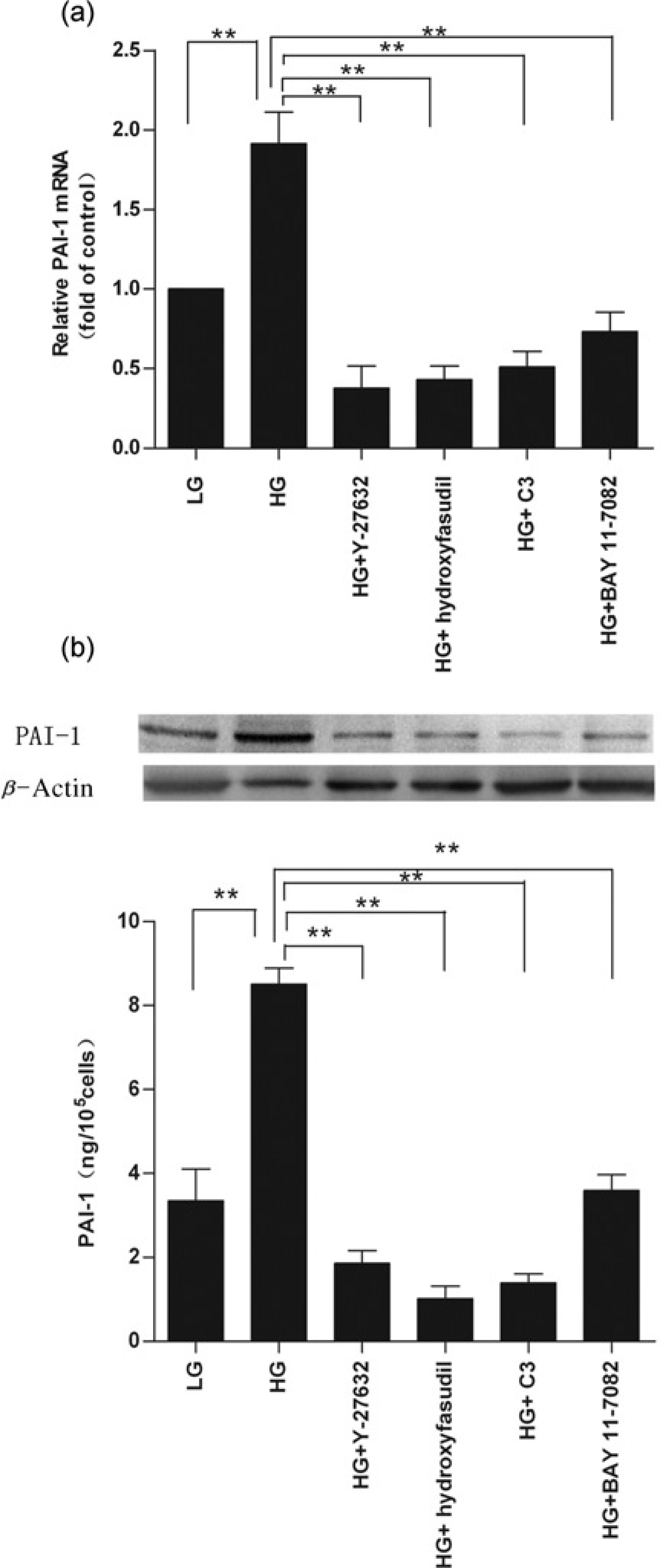
BAY 11-7082, a specific inhibitor of NF-κB by selectively and irreversibly inhibiting IΚBα phosphorylation, at 5 μmol/L also demonstrated significant suppression of high glucose-induced increases of PAI-1 mRNA and protein expressions (all P < 0.01; Figures 3a and b). Treatment with BAY 11-7082 did not affect cell viability (data not shown).
Effects of simvastatin and atorvastatin on high glucose-induced RhoA activation
RhoA was activated by high glucose stimulation as shown by the elevated activated GTP-bound configuration (Figure 4). Treatment with simvastatin (10 μmol/L) or atorvastatin (10 μmol/L) significantly inhibited RhoA activation as shown by GTP-RhoA/total RhoA ratios (Figure 4).
Statins inhibited high glucose-induced RhoA activation. Cardiac microvascular endothelial cells were pretreated with 10 μmol/L statins for 30 min before high glucose stimulation in the presence of statins for 24 h. (a) Western blotting of GTP-RhoA and total RhoA. (b) Ratios of GTP-RhoA/ total RhoA. LG, low glucose (5.7 mmol/L); HG, high glucose (23 mmol/L); PA-1, plasminogen activator inhibitor-1. Data are expressed as means ± SD (n = 3). **P < 0.01 versus HG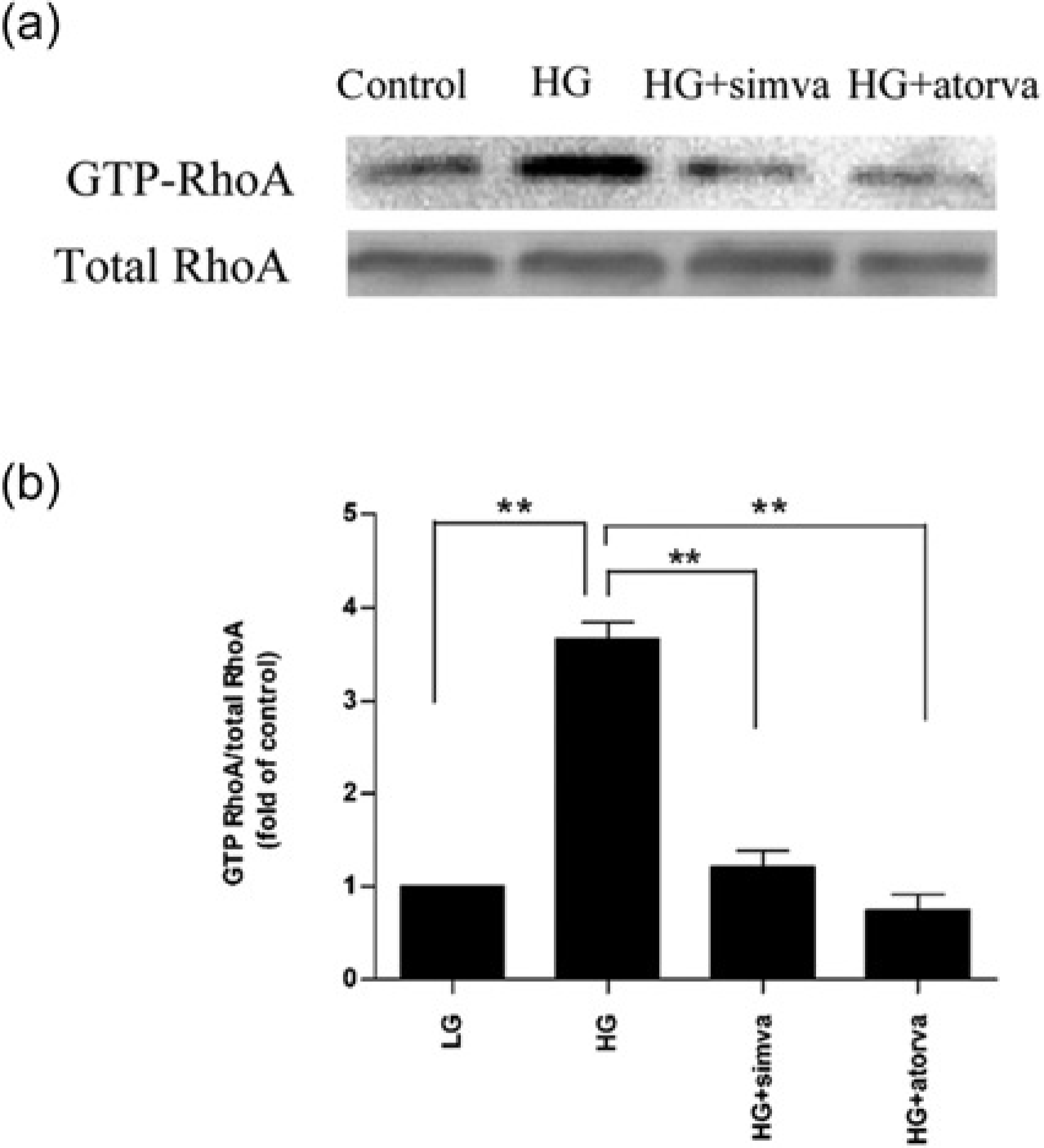
Effects of simvastatin and atorvastatin on high glucose-induced NF-κB activity
High glucose (23 mmol/L) induced IΚBα degradation from five minutes until 60 min after stimulation, and IΚBα degradation returned to the basal level at 120 min (Figure 5). When CMECs were in the presence of 10 μmol/L simvastatin or atorvastatin, the high glucose-induced IΚBα degradation was completely blocked (Figure 5). As indicated by EMSA, simvastatin and atorvastatin suppressed high glucose-induced NF-κB activation. The effects were similar to Rho/ROCK inhibitor (Figure 6a).
Inhibitory effects of simvastatin and atorvastatin on high glucose-induced IΚBα degradation. Cardiac microvascular endothelial cell were cultured in high glucose (23 mmol/L) for various time periods with or without simvastatin (10 μmol/L) or atorvastatin (10 μmol/L)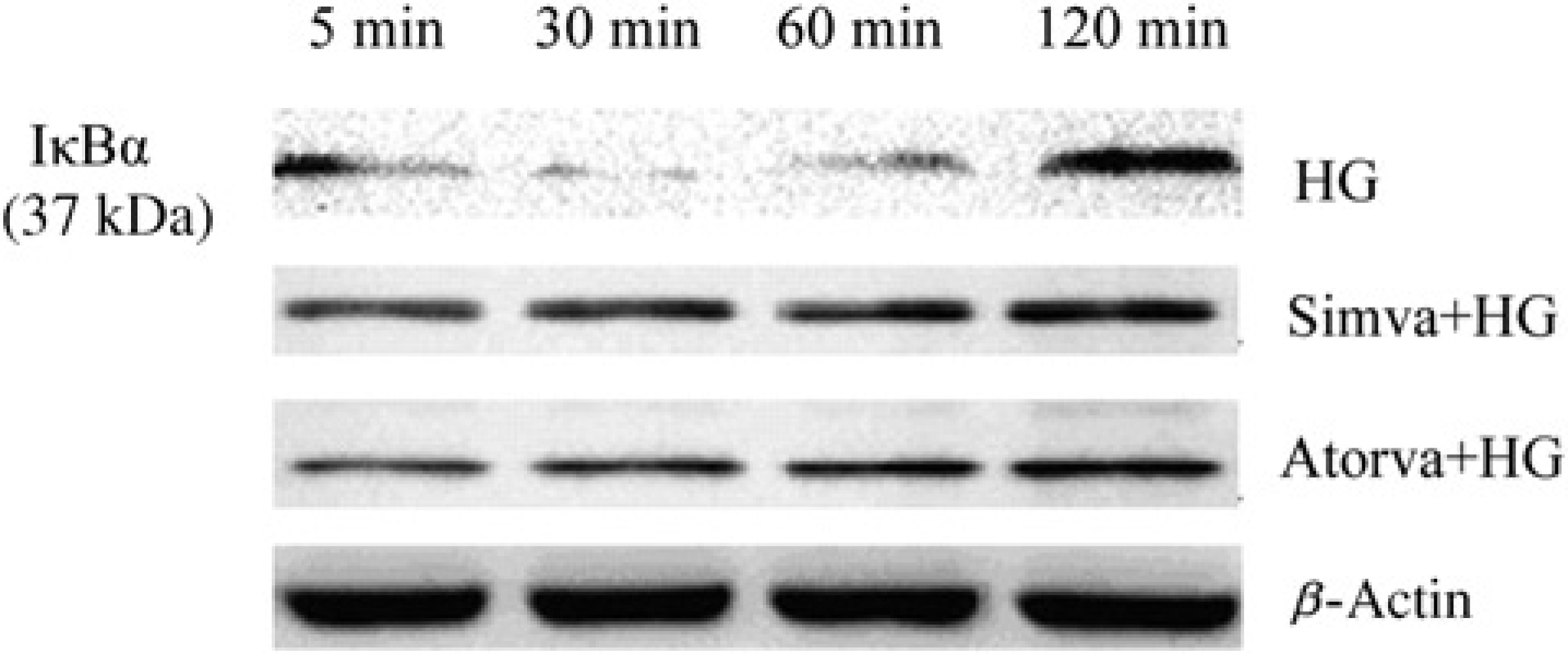
Effects of statins and RhoA/ROCK inhibitors on high glucose-induced DNA-binding activity to the nuclear factor κB (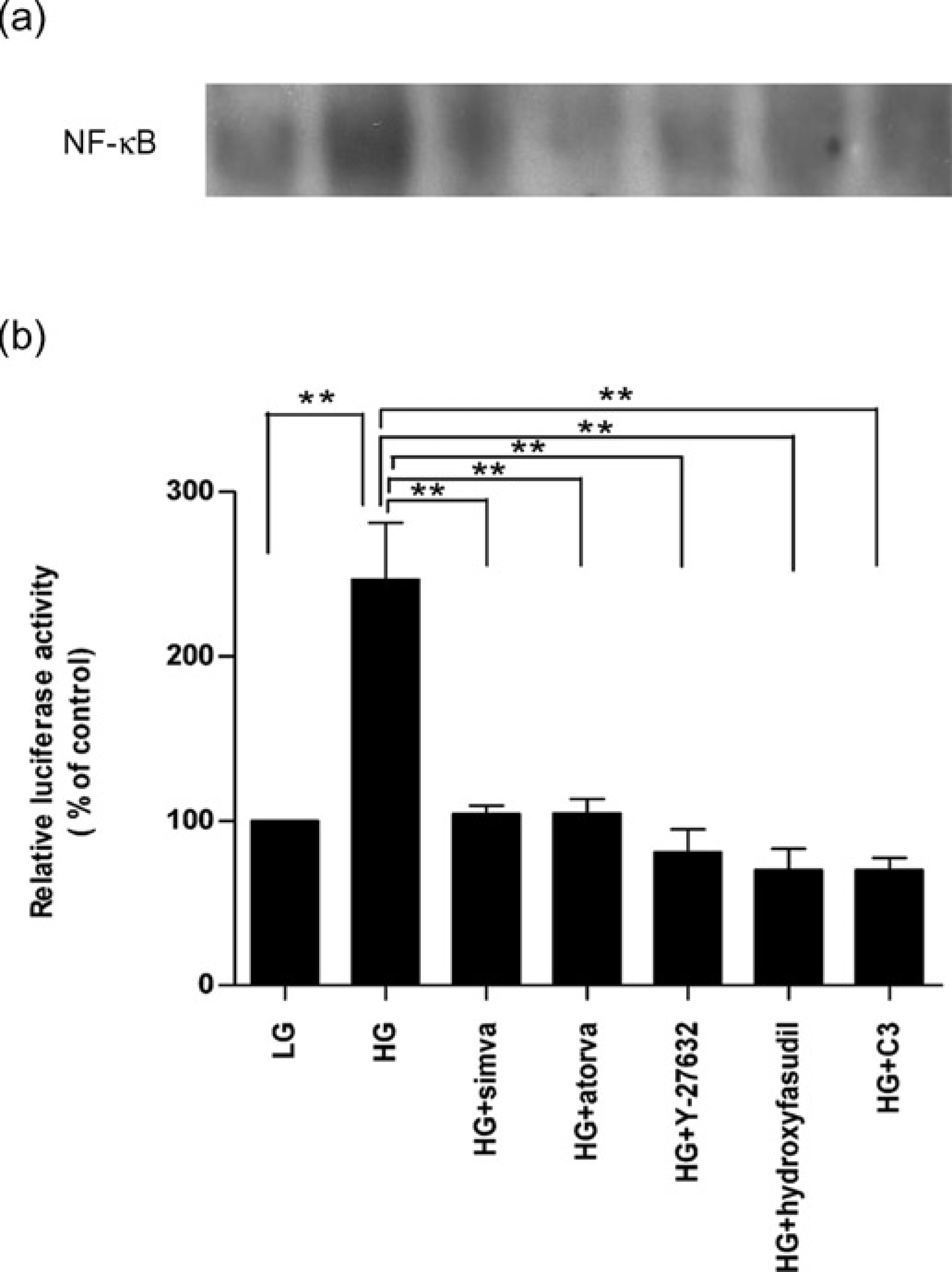
Consistently, high glucose (23 mmol/L) induced the NF-κB-dependent transcriptional activity by over two-fold, compared with control low glucose treatment, and this activation was completely inhibited by simvastatin and atorvastatin to the similar robust effects of the specific RhoA/ROCK inhibitors Y-27632, hydroxyfasudil and C3 exoenzyme, respectively (all P < 0.01; Figure 6b).
Discussion
Endothelial PAI-1 production can be induced by numerous factors such as tumor necrosis factor α, 20 thrombin, 21 angio-tensin II, 22 transforming growth factor, 23 etc. Increased PAI-1 production induced by high glucose was previously observed in human umbilical vein endothelial cells (HUVECs), 24 bovine aortic endothelial cells (BAECs) 25 and human saphenous vein endothelial cells (HSVECs). 26 Our present study suggested that high glucose also increased PAI-1 production in CMECs. The glucose-induced production of PAI-1 was in a concentration- and time-dependent manner (Figure 1). In BAECs, it has been shown that high glucose induces PAI-1 secretion starting at six hours, reaches the peak at 12 h and recovers at 24 h of stimulation; 25 whereas in HSVECs, high glucose at the same concentration induces PAI-1 production to peak at 72 h of stimulation. 26 Our results show that PAI-1 mRNA and protein levels began to rise at six hours of stimulation, PAI-1 mRNA peaked at 12–24 h and PAI-1 protein level still kept increasing until 72 h. Together, these different patterns indicate cell-specific inductions of PAI-1 responding to hyperglycemia.
Many clinical studies have suggested numerous effects of statins on patients with heart failure, type 2 diabetes and lipid dysfunction that are often accompanied by an improvement of the fibrinolytic system, including normalization of plasma PAI-1 levels.27,28 Statins have been demonstrated to suppress the production of PAI-1 in HUVECs and HSMECs, 29 adipocytes, 30 monocytes 11 and vascular smooth muscle cells (VSMC),17,31 but not in HepG2 cells. 29 In our experiments, simvastatin and atorvastatin showed robust inhibitory effects on high glucose-induced PAI-1 mRNA and protein expressions when the concentrations of statins increased up to 10 μmol/L (Figure 2). The effects can be reversed by MVA (100 μmol/L) or GGPP (10 μmol/L) but not FPP (10 μmol/L) suggest that such effects are through MVA pathway unrelated to their lipid-lowering action. These results were consistent with previous reports in endothelial cells of other origins.17,29 These data indicate that statins improve the fibrinolytic function of the endothelium by lowering PAI-1 levels.
We found that the activated form of RhoA (GTP-RhoA) was significantly increased after stimulation with high glucose treatment, which was consistent with a previous study. 26 We also observed that the high glucose-induced elevation of PAI-1 protein expression was significantly suppressed by the Rho-specific inhibitor C3 exoenzyme and ROCK inhibitors Y-27632 and hydroxyfasudil (Figure 3). These data indicate that the increased expression of PAI-1 stimulated by high glucose may be via activation of the Rho/ROCK pathway in CMECs. We further observed that simvastatin and atorvastatin significantly suppressed RhoA activation (Figure 4). It is logical to conclude that statins downregulate the synthesis of PAI-1 via inactivation of RhoA. Since it is GTP-RhoA and not total RhoA that activates downstream signals, our method using a GST pulldown assay to measure the GTP bound form of RhoA was superior to previous investigations that measured membrane RhoA to determine the status of RhoA activation. This methodology may be one of the reasons that there are conflicting results in previous studies. The effects of simvastatin (10 μmol/L) and atorvastatin (10 μmol/L) inhibition of PAI-1 expression were similar to the effects of Rho/ROCK inhibitors but the mechanism of statins may be different from that of RhoA/ROCK inhibitors, since the RhoA/ROCK inhibitors compete the ROCK binding site with ATP 32 whereas statins suppress the synthesis of iso-prenoids including GGPP, which is known to be the major donor of hydrophobic isoprenoids to Rho, 33 may lead to indirect suppression of the RhoA/ROCK signaling pathway.
In unstimulated cells, NF-κB is sequestered in the cytoplasm and inhibited by members of the IΚB family, mainly including IΚBα and IΚBβ. Degradation of IΚBα plays a critical role in translocation of NF-κB into the nucleus. Once activated by various stimuli such as proinflammatory cytokines, growth factors or chemotatic factors, IΚBα is normally phosphorylated at Ser32/36 by IΚB kinase complexes and rapidly degraded. This leads to release of NF-κB from IΚB and its translocation to the nucleus, which in turn initiates gene transcription by binding of NF-κB to specific promoter elements. 34 The level of IΚBα is rapidly decreased and then recovered during the course of NF-κB activation. Our present study showed that decreased IΚBα and NF-κB activation after high glucose stimulation (Figures 5 and 6) was consistent with the results of previous studies. 17 C3 exoenzyme, Y-27632 and hydroxyfasudil inhibited the increased activity of NF-κB transcription induced by high glucose, suggesting that inhibition of the RhoA/ROCK pathway may cause suppressed NF-κB activation. On the other hand, high glucose-induced PAI-1 expression was blocked by the NF-κB inhibitor BAY 11-7082 (Figure 3), suggesting that the NF-κB signaling pathway is involved in the production of PAI-1.
RhoA has been shown to be the upstream regulator of NF-κB in tight junctions of brain microvascular cell lines. 35 Consistently, inhibition of ROCK may lead to inhibition of NF-κB activation, 36 whereas RhoA activation may lead to activation of NF-κB via several downstream pathways. 37 Inhibition of the RhoA/ROCK pathway blocked the expression of the NF-κB reporter gene in BAECs cultured in high concentration of glucose, which suggested that high glucose induced PAI-1 expression through the NF-κB-dependent signaling pathway via activation of the Rho/ROCK signaling pathway. 25 Our investigation in CMECs was in agreement with these studies. Furthermore, we observed that simvastatin and atorvastatin inhibited IΚBα degradation and NF-κB activation. In addition, inhibition of RhoA/ROCK suppressed NF-κB activation. These results suggest that statins inhibit high glucose-induced PAI-1 expression in CMECs, which is partially mediated by NF-κB activation through the RhoA/ROCK pathway, and RhoA is a positive upstream regulator of NF-κB.
Simvastatin and atorvastatin are naturally occurring and synthetic statins, respectively, but their effects on PAI-1 production and its signaling pathway in CMECs induced by high glucose are very similar in the present study. A cyto-toxic study of statins on hepatic cells has indicated that atorvastatin (1–30 μmol/L for 24 h) caused a concentration-dependent cell injury, 38 whereas another study reported that 5 μmol/L statins caused a significant LDH leakage in VSMC. 29 However, we found that simvastatin and atorvastatin at 0.1–10 μmol/L did not influence cell viability; but 30 μmol/L statins significantly enhanced LDH leakage, which indicated obvious cell injury. This difference may due to cell type-specific tolerance of statins. We used 10 μmol/L statins in the remaining experiments so that the concentration was the same as most previous studies. We used 10 μmol/L statins in the vitro experiments which was supraphysiological compared with the doses of statins in patients under treatment; however, it should be noted that the concentration was comparable to previous studies analyzing the effects of statins on different cell signal pathway.17,29
An earlier study reported that three different NF-κB inhibitors, parthenolide, BAY 11-7082 and SN50, had little effect on PAI-1 production in HUVECs; however, after high glucose stimulation for 12 h, significant inhibition was observed even at low doses. 25 Y-27632 inhibited NF-κB activation in BAECs cultured in high glucose but not in low glucose. 25 These data suggest that in addition to stimuli, the cell basal state may determine whether RhoA/ROCK and NF-κB signaling pathways are activated or not. Interestingly, lovastatin has been found to activate IKK/NF-κB via inhibiting the RhoA/ROCK pathway. 39 In addition, simvastatin has been shown to activate NF-κB, whereas interfering siRhoA has been shown to upregulate NF-κB-dependent transcriptional activity. 40 The inhibitory effect of statins on NF-κB signaling has been shown in monocytes, hepatocytes and endothelial cells after stimulation but not under cell basal conditions. 30 These discrepancies between the previously reported effects of statins and our data on RhoA/ROCK and NF-κB signaling may be attributable to various factors such as different stimuli, activation levels of the cells, cell types and sources, categories and doses of statins.
There are many other potential signaling pathways, including RAC, 14 MEK, 30 P38MAPK 13 and PI3K/AKT, 3 through which statins may regulate PAI-1 production. Statins have been found to diminish procoagulant activity by influencing different stages of the coagulation cascade such as upregulating tissue-type plasminogen activator (t-PA) 29 and suppressing tissue factor. 21 The effects of statins on balance between t-PA and PAI-1 expression in hyperglycemia and the kinetic mechanism remain to be investigated in our future studies. Considering that the pleiotropic effects of statins are more than the inhibitive effect of RhoA, statins may be superior to the RhoA kinase inhibitors fasudil and hydroxyfasudil, which are widely used in the clinic now. Although some evidence has indicated that a combination therapy of a statin and a RhoA-kinase inhibitor could exert more effective cardiovascular protection in rats, 14 it should be verified in large clinical trials, and the side-effects should be thoroughly investigated.
In summary, our study demonstrated that both simvastatin and atorvastatin suppressed PAI-1 production in CMECs induced by high glucose via inhibition of the RhoA/ROCK–NF-κB-dependent signaling pathways. The RhoA/ROCK–NF-κB signaling pathway may play an important role in microvascular thrombosis in hyper-glycemia. Given their pleiotropic effects, such as cholesterol lowering and anti-inflammatory, statins may have significant clinical implications, especially for metabolic syndrome and diabetes with microvascular complications.
Footnotes
Acknowledgements
This study was supported by the Jiangsu Provincial Outstanding Medical Academic Leader program (No: LJ201140) and the National Natural Science Foundation of China (No: 81070139).