
Abstract
Intimal hyperplasia results in significant morbidity and mortality following vascular intervention. Both platelets and elevated homocysteine have been implicated in the development of intimal hyperplasia. We previously demonstrated that a locally applied antiplatelet agent decreases the development of intimal hyperplasia. We were therefore interested in a systemic antiplatelet agent, clopidogrel. We hypothesized that clopidogrel would decrease platelet aggregation and activity and intimal hyperplasia. Male Sprague-Dawley rats underwent carotid endarterectomy (CEA) and treatment with either placebo or varying regimens of clopidogrel, including chronic, pre-CEA bolus, chronic plus pre-CEA bolus, and chronic plus post-CEA bolus; a homocystine diet was used to elevate both plasma homocysteine and the degree of intimal hyperplasia. Platelet aggregation, platelet activity, and intimal hyperplasia were then assessed. Platelet aggregation was not decreased with chronic clopidogrel; however, it was decreased with pre-CEA bolus clopidogrel. Similarly, platelet activity was not inhibited by chronic clopidogrel but was inhibited by pre-CEA and chronic plus pre-CEA bolus clopidogrel. Neither chronic, pre-CEA bolus, chronic plus pre-CEA bolus, nor chronic plus post-CEA bolus clopidogrel resulted in a decrease in intimal hyperplasia. Although pre-CEA bolus clopidogrel resulted in a decrease in both platelet aggregation and activity, it was unable to decrease the development of intimal hyperplasia at any dose. Additional factors must therefore contribute to the pathologic development of intimal hyperplasia.
Intimal hyperplasia is a significant problem, frequently occurring throughout the vascular tree after therapeutic manipulation. Thirty to forty percent of percutaneous transluminal coronary angioplasties develop restenosis within 3 to 4 months. 1 Thirty percent of peripheral vein grafts develop intimal hyperplasia within the first year, contributing to 20 to 40% of first-year graft failures. 2 Furthermore, 10 to 20% of patients undergoing carotid endarterectomy (CEA) develop greater than 50% restenosis attributable to intimal hyperplasia 3 ; 2% of these patients will develop > 80% restenosis, resulting in an ipsilateral stroke rate of 5.6%. 4 The primary patency rate at 1 year following angioplasty and stenting of the superficial femoral artery is as low as 22%. 5 Given the magnitude of this problem, we felt that it was necessary to investigate the contributors to intimal hyperplasia development and attempt to interfere with its pathologic formation.
Many cellular components and inflammatory mediators interact after vascular wall injury whose actions collectively result in the development of intimal hyperplasia. Injury to the vessel wall removes the protective endothelial cell layer, exposing the underlying connective tissue. Exposure of the subendothelium results in platelet adhesion and activation. On activation, platelets degranulate, releasing multiple inflammatory mediators, including platelet-derived growth factor, von Willebrand's factor, and adenosine diphosphate (ADP). Platelets then aggregate through various mechanisms, which include binding adhesive glycoproteins via their glycoprotein IIb/IIIa surface receptor, the predominant platelet receptor for fibrinogen binding. ADP, through its binding of the P2Y12 platelet surface receptor, is an indirect activator of glycoprotein IIb/IIIa, thus promoting platelet aggregation. Additional aggregation with subsequent activation of platelets further contributes to the injurious cascade. Platelet-secreted growth factors stimulate the migration and proliferation of vascular smooth muscle cells, which produce and secrete extracellular connective tissue matrix. This abnormal migration, proliferation, and secretion define intimal hyperplasia, resulting in significant restenosis, which increases patient morbidity and mortality.
Clopidogrel is frequently used in the clinical setting as an antiplatelet agent. Clopidogrel irreversibly binds the platelet P2Y12 surface receptor, whose normal binding of ADP is necessary for glycoprotein IIb/IIIa activation and thus platelet aggregation. Glycoprotein IIb/IIIa contributes to platelet aggregation. 6 Clopidogrel irreversibly inhibits the ADP platelet surface receptor, P2Y12, blocking platelet aggregation. 7 Multiple studies demonstrate a significant decrease in platelet aggregation to injured blood vessel walls with clopidogrel treatment. 1,8,9
Homocysteine may also affect platelet activity and the development of intimal hyperplasia. Homocysteine, a non–protein-forming amino acid, is an independent risk factor for cardiovascular disease. The proliferation of vascular smooth muscle cells, which has been implicated as the response to injury resulting in the development of intimal hyperplasia, is increased by homocysteine. 10 It may also result in hyperactivation of platelets. 11 In addition, intimal hyperplasia increases proportionately with increasing levels of homocysteine in a rat CEA model. 12
Given the possible relationship between clopidogrel and its inhibition of platelet aggregation, the probable relationship between platelets, vascular smooth muscle cell proliferation, and intimal hyperplasia, in addition to the morbidity of restenosis, we investigated the effect of clopidogrel on platelet aggregation, platelet activity, and intimal hyperplasia development. Using an animal model of CEA, which produces significant arterial injury, resulting in restenosis, we hypothesized that clopidogrel would decrease platelet aggregation, platelet activity, and intimal hyperplasia in both control and hyperhomocysteinemic environments.
Methods
Platelet Aggregation, Platelet Activity, and Intimal Hyperplasia
Male Sprague-Dawley rats (N = 107) weighing ≥450 g body weight were placed into one of three groups: group 1 evaluated platelet aggregation (n = 28) on the carotid surface 3 hours after CEA, group 2 measured platelet activity as P2Y12 receptor units (n = 26), and group 3 measured the development of intimal hyperplasia formation (n = 53) 2 weeks after CEA.
In group 1, platelet aggregation rats were subdivided into three groups that included administration of the following treatments: placebo (n = 9), chronic clopidogrel at 1 mg/kg/d for 5 days prior to CEA (n = 12), or 4.3 mg/kg pre-CEA bolus 4 hours prior to endarterectomy (n = 7). Platelet aggregation rats were reanesthetized 3 hours after CEA. The endarterectomized arteries were harvested and opened longitudinally along the suture line, exposing the endarterectomized area. The arteries were then fixed in a 4% glutaraldehyde solution and then postfixed with osmium tetroxide, dehydrated in a graded alcohol series, critical point dried with carbon dioxide (1,072 psi and 31.1°C), and coated with gold palladium. They were then placed in a scanning electron microscope (JEOL JSM 5410, JEOL, Peabody, MA, USA). The endarterectomized areas were scanned at ×2,000 magnification, and photographic images were obtained. The photographic images were matched and arranged in a collage, allowing visualization of a greater area than was possible with a single field of the scanning electron microscope monitor. Once the collage of photographs was assembled, it was covered with a transparent overlay grid. The same overlay grid was used for all specimen photographs; thus, the same number of grid squares was used to count the total number of platelets in each photograph. One hundred sixteen squares were the maximum number of squares that could be consistently counted on all of the photographed collages. Platelet counts were performed by two blinded observers and expressed as platelets per high-power field.
In group 2, platelet activity rats received either placebo (n = 8) or clopidogrel for 5 days at one of three clopidogrel dosing regimens. Rats had approximately 2 mL of blood collected into vacuette tubes (9NC coagulation sodium citrate 3.2%) for analysis on an Ultegra device (Accumetrics, San Diego, CA). Eighteen rats received clopidogrel. Chronic clopidogrel (1 mg/kg/d × 5 days) was administered to four rats, pre-CEA bolus clopidogrel (4.3 mg/kg) was administered to seven rats, and chronic plus pre-CEA bolus clopidogrel (1 mg/kg/d × 4 days plus 4.3 mg/kg bolus on the fifth day) was administered to seven rats. Four hours after rats received their last treatment, blood was collected and measured on the Ultegra device. By assessing the degree of platelet binding to fibrinogen-coated beads, thus approximating the degree of occupation of the P2Y12 platelet surface receptor by clopidogrel, the Ultegra device was used to measure platelet activity. The degree of platelet activity was then expressed as P2Y12 receptor units. A greater occupancy of the receptor by clopidogrel resulted in a lesser degree of platelet binding and thus smaller P2Y12 receptor unit numbers for platelet activity.
In group 3, intimal hyperplasia rats were subdivided into two dietary groups: control diet rats (n = 33) and homocystine diet rats (4.5 g/kg
Plasma Homocysteine Measurements
All plasma homocysteine laboratory values were determined by the Clinical Pathology Department at the Central Arkansas Veterans Healthcare Facility, Little Rock, AR. The plasma homocysteine level was measured using a thiol-specific fluorogenic labeling reagent, and the thiols were separated by a reverse-phase high-pressure liquid chromatography method using fluorescence detection. 14
Rat CEA
A detailed description of the rat CEA was reported by Southern and colleagues. 15 In brief, rat endarterectomy involved exposure of the rat common carotid artery followed by arteriotomy. Following arteriotomy, a 1 × 0.5 mm section of normal endothelium was then removed. The intimectomized or endarterectomized artery was then closed in a running fashion for later harvest.
Statistics
Data were analyzed as mean ± SE by analysis of variance and simple regression analysis using StatView for Windows, version 5.0 (SAS Institute Inc. Cary, NC).
Results
When evaluating platelet aggregation, chronic clopidogrel (1 mg/kg/d × 5 days) did not effectively inhibit aggregation when compared with placebo (Figure 1). This is illustrated inFigure 2, in which the endarterectomized surface of a rat carotid artery treated with chronic clopidogrel appears to be nearly identical to a rat carotid artery treated with placebo. In contrast, pre-CEA bolus clopidogrel (4.3 mg/kg) significantly reduced platelet aggregation where aggregation was decreased to only six platelets per high-power field, a reduction of 82% when compared with rats receiving placebo (seeFigure 1). The absence of platelet aggregation on the endarterectomized surface of an artery treated with pre-CEA bolus clopidogrel is illustrated inFigure 2.
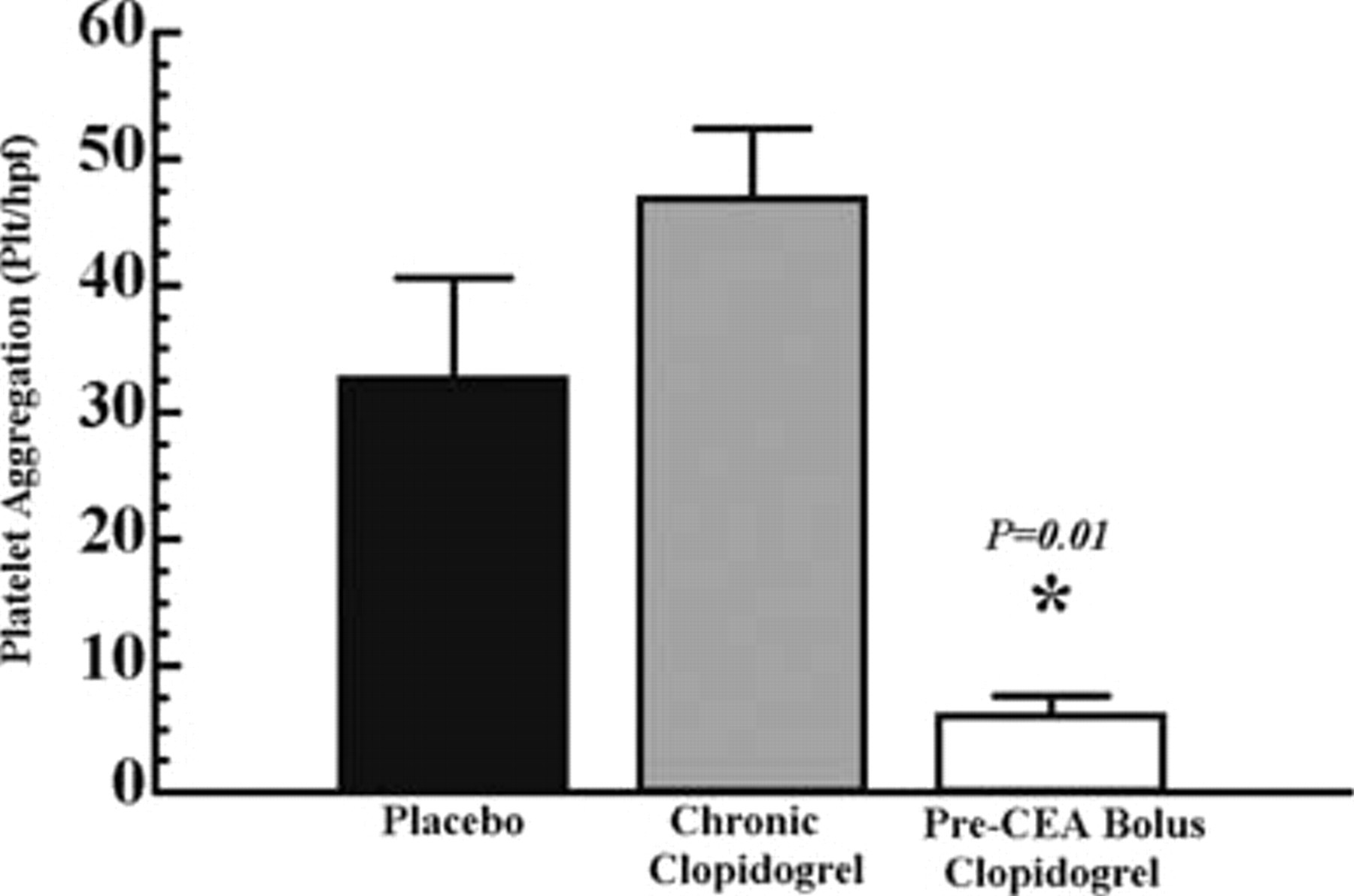
Platelet aggregation on the endarterectomized rat carotid artery 3 hours after endarterectomy in rats receiving a control diet and placebo, chronic clopidogrel, or pre–carotid endarterectomy (CEA) bolus clopidogrel. Platelet aggregation is defined as the number of platelets per high-power field using scanning electron microscopy. Chronic clopidogrel was administered at 1 mg/kg/d × 5 days prior to endarterectomy. Differences in platelet aggregation were not significant when comparing placebo and chronic clopidogrel (p = .10). Pre-CEA bolus clopidogrel (4.3 mg/kg) given 4 hours prior to endarterectomy resulted in a significant decrease in platelet aggregation when compared with placebo (p = .01), designated by an asterisk. Data are represented as mean ± SEM.
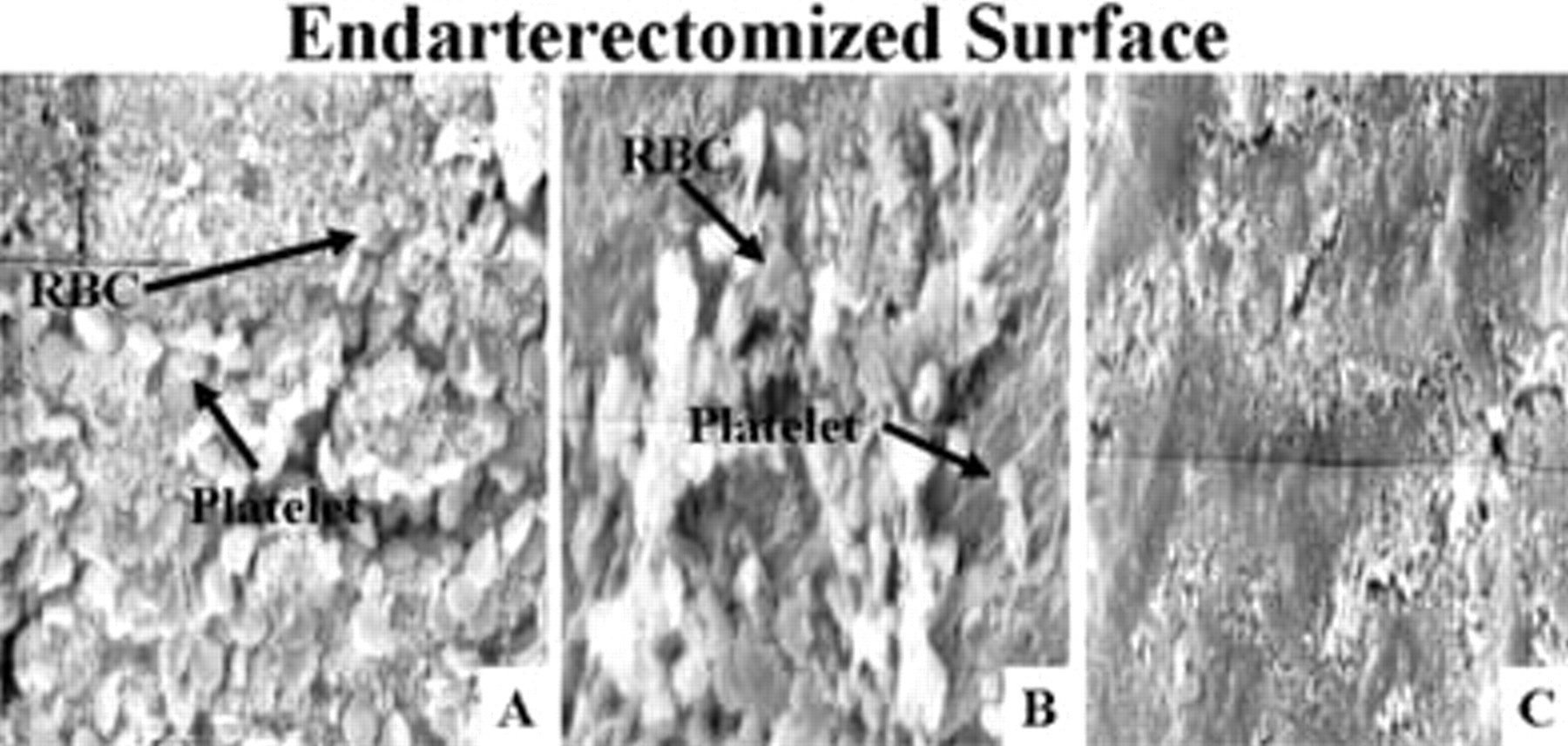
Scanning electron microscopy photographs of the intimectomy site on the common carotid artery in rats receiving a control diet and treated with placebo, chronic clopidogrel, or pre–carotid endarterectomy (CEA) bolus clopidogrel. Chronic clopidogrel was administered at 1 mg/kg/d × 5 days prior to endarterectomy. A represents placebo, whereas B represents chronic clopidogrel. Note the similarity between A and B showing minimal difference in platelet aggregation between arteries treated with placebo or chronic clopidogrel. A pre-CEA bolus dose of clopidogrel (4.3 mg/kg) administered 4 hours prior to endarterectomy resulted in a significant decrease in platelet aggregation when compared with placebo. C represents pre-CEA bolus clopidogrel. Note the significant difference in platelet aggregation between A and C. Original magnification ×2,000. RBC = red blood cells.
Although chronic clopidogrel was ineffective in decreasing platelet activity (Figure 3), both chronic plus pre-CEA bolus clopidogrel and pre-CEA bolus clopidogrel significantly decreased platelet activity. Chronic plus pre-CEA bolus clopidogrel (1 mg/kg/d × 4 days plus 4.3 mg/kg on day 5, 4 hours prior to endarterectomy) resulted in a 60% reduction in platelet activity when compared with rats receiving placebo (seeFigure 3). Pre-CEA bolus clopidogrel (4.3 mg/kg 4 hours prior to endarterectomy) decreased platelet activity to 50% of platelet activity observed in placebo-treated rats (seeFigure 3).
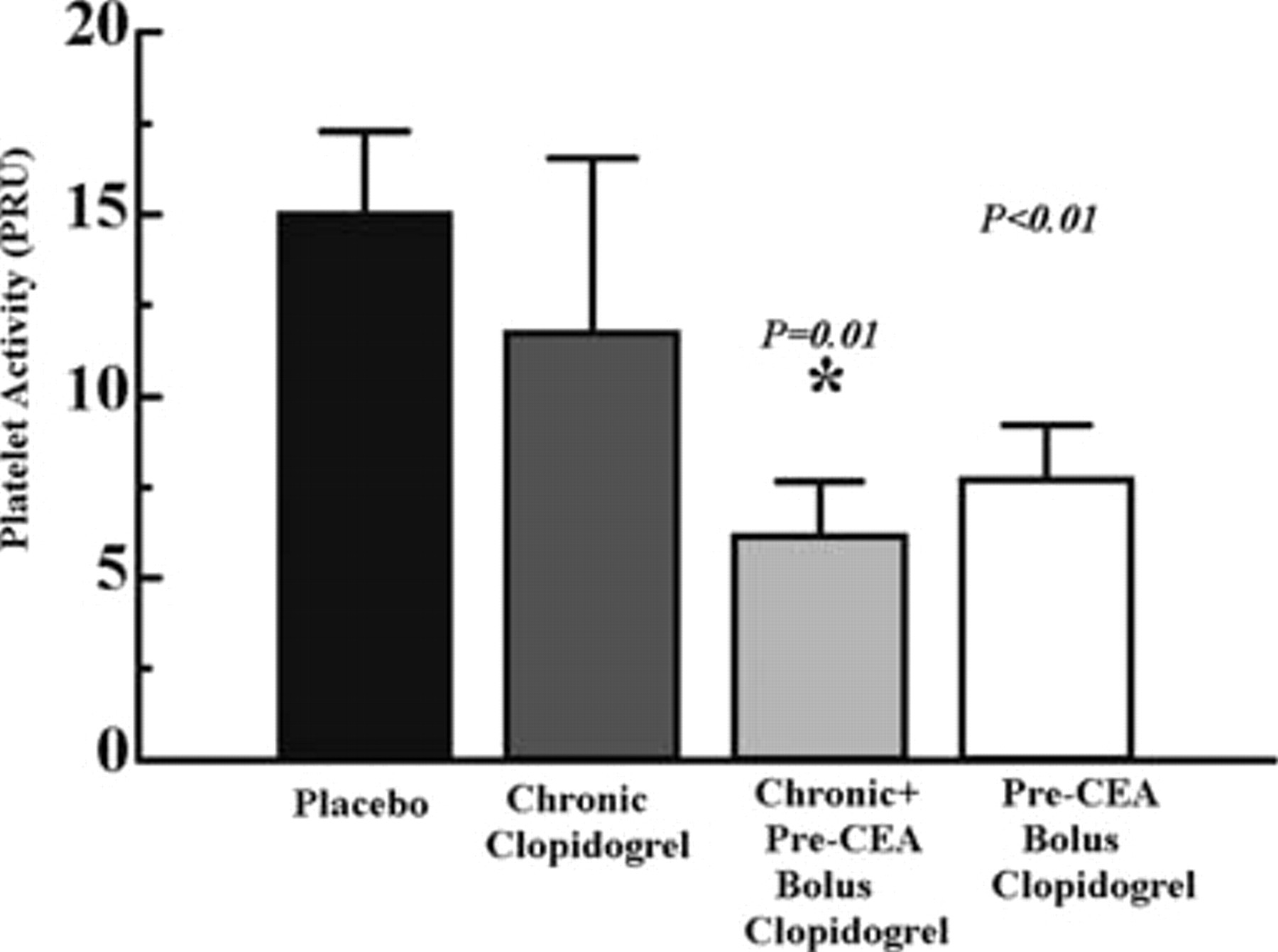
Platelet activity as P2Y12 receptor units (PRUs) in rats receiving a control diet and placebo, chronic clopidogrel, chronic plus pre–carotid endarterectomy (CEA) clopidogrel, or pre-CEA bolus clopidogrel. The degree of platelet activity was expressed as PRUs. Chronic clopidogrel was administered at 1 mg/kg/d × 5 days. The results were not significantly different (p = .38) when compared with placebo. In the chronic plus pre-CEA bolus group, chronic clopidogrel was administered at 1 mg/kg/d × 4 days. A pre-CEA bolus (4.3 mg/kg) was then administered on day 5 4 hours prior to blood collection for measurement of platelet activity. In comparison with placebo, the results were significantly different (p = .01), designated by an asterisk. Pre-CEA bolus clopidogrel (4.3 mg/kg) was administered 4 hours prior to blood collection for measurement of platelet activity. The results were significantly different (p = .01) in comparison with placebo, designated by a dagger. Data are represented as mean ± SEM.
Clopidogrel was ineffective in decreasing intimal hyperplasia at any dosing regimen (Figure 4) irrespective of its effect on platelet aggregation and activity. In rats receiving a homocystine-supplemented diet, although the degree of intimal hyperplasia was significantly increased when compared with rats receiving the control diet, 72.3% versus 30.6%, respectively, intimal hyperplasia response was again unchanged when chronic plus post-CEA bolus clopidogrel was administered when compared with placebo (Figure 5). Plasma homocysteine values were not significantly different between the placebo and the chronic + post-CEA bolus groups (p = not significant).
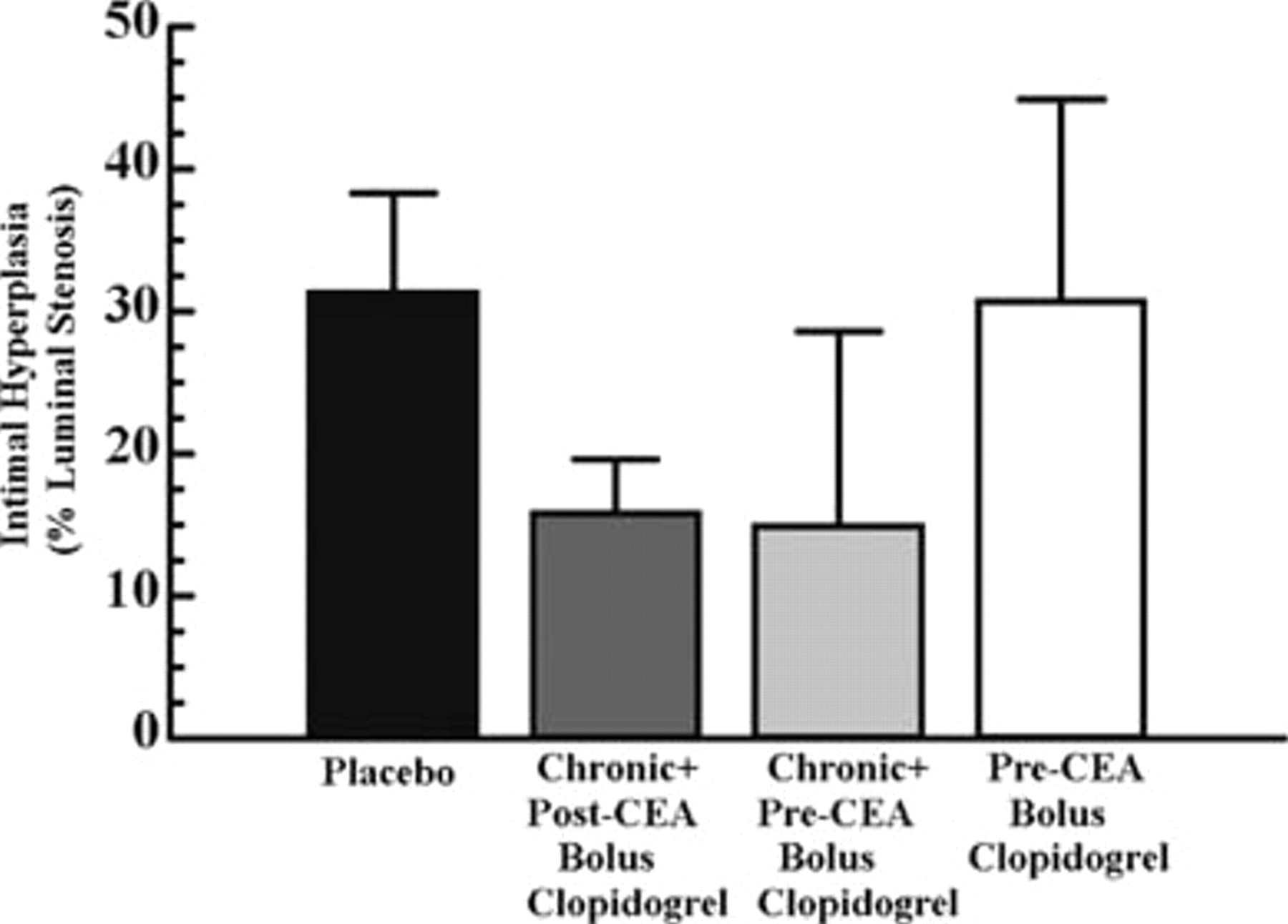
Intimal hyperplasia in rats receiving a control diet and placebo versus chronic plus post–carotid endarterectomy (CEA) bolus clopidogrel, chronic plus pre-CEA bolus clopidogrel, or pre-CEA bolus clopidogrel. Intimal hyperplasia 2 weeks after endarterectomy is represented as percent luminal stenosis of the endarterectomized carotid artery. In chronic plus post-CEA bolus, chronic clopidogrel was administered at 1 mg/kg/d × 5 days. A post-CEA bolus was then administered at 4.3 mg/kg 4 hours after endarterectomy. In comparison with placebo, the results were not significantly different (p = .24). The chronic plus pre-CEA bolus group received chronic clopidogrel at 1 mg/kg/d × 4 days. Pre-CEA bolus clopidogrel (4.3 mg/kg) was then administered on day 5 4 hours prior to endarterectomy. The results were not significantly different (p = .96) when compared with placebo. Pre-CEA bolus clopidogrel was administered at 4.3 mg/kg 4 hours prior to endarterectomy. Again, the results were not significantly different (p = .96) in comparison with placebo. Chronic clopidogrel (1 mg/kg/d) was maintained after endarterectomy until sacrifice 2 weeks later in all clopidogrel-treated groups. Data are represented as mean ± SEM.
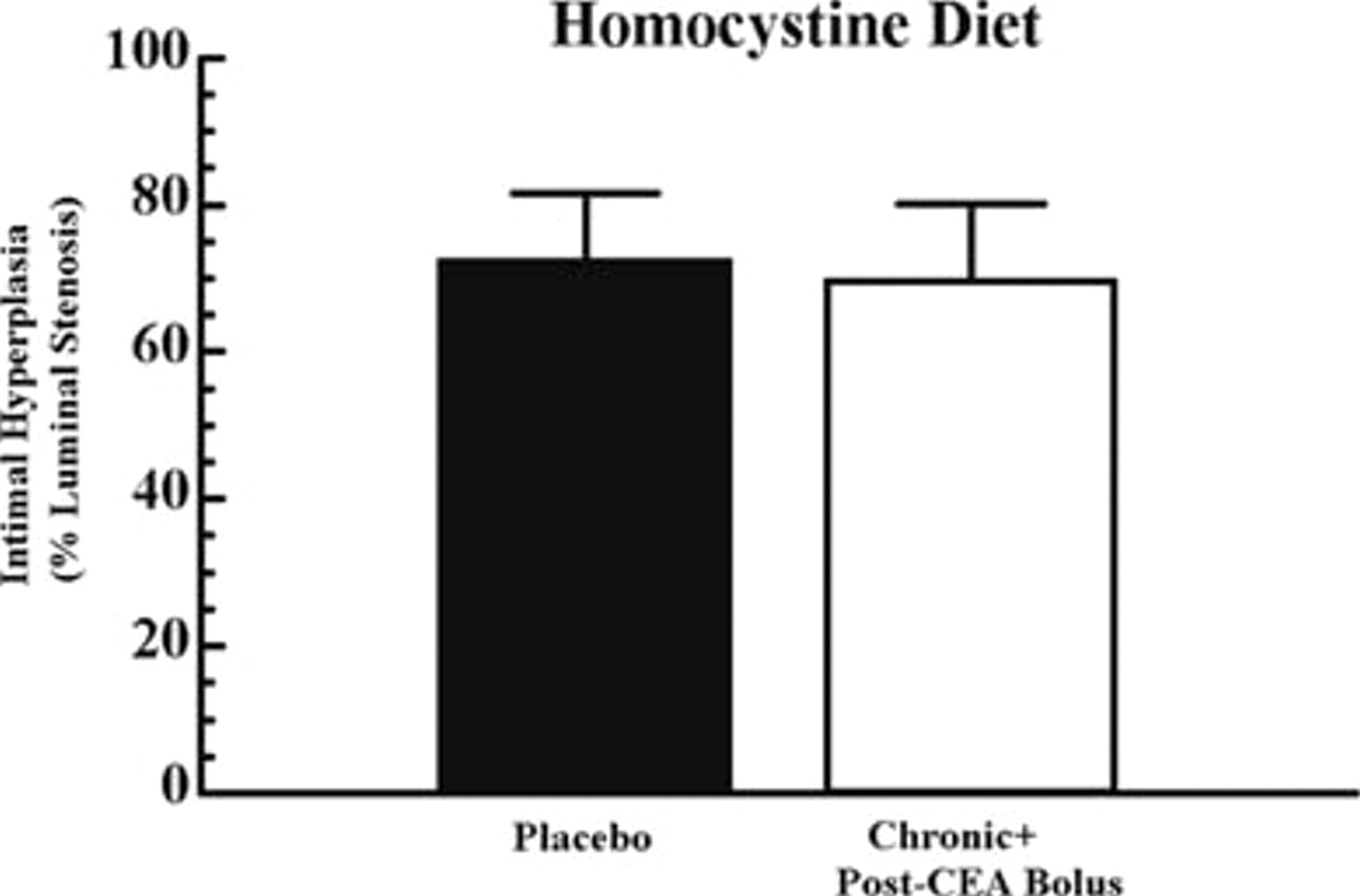
Intimal hyperplasia in rats receiving homocystine-supplemented diets and placebo versus chronic plus post–carotid endarterectomy (CEA) bolus clopidogrel. Rats were given 4.5 g/kg
Discussion
In our rat model of CEA, clopidogrel did not inhibit the development of intimal hyperplasia at any dosing regimen and only decreased platelet aggregation and activity at the preprocedural bolus dose. We used a rat CEA model as opposed to the more commonly used balloon injury model. We feel that the CEA model is more pathologically equivalent in promoting platelet aggregation and activity at a site of vascular injury. As opposed to commonly used models that stretch the artery, our model uses a suture line, which should be evaluated when administering platelet aggregating and/or activating drugs. Because ADP is a prominent promoter of platelet aggregation, we chose clopidogrel, a highly effective platelet ADP receptor blocker, to investigate inhibition of platelet aggregation and activity and possibly intimal hyperplasia. Given that clopidogrel often is not administered prior to vascular intervention, we first investigated a post-CEA bolus of clopidogrel in the hope that it would significantly decrease platelet aggregation and activity and intimal hyperplasia. We did not observe a decrease in platelet aggregation or activity; thus, we were not surprised when intimal hyperplasia was not decreased. We therefore investigated a pre-CEA bolus dose both with and without pre-CEA chronic clopidogrel. Comparable bolus 16 and chronic 17 doses have been investigated. Although we did find a decrease in platelet aggregation and activity, intimal hyperplasia response was not affected. Thus, one may speculate that clopidogrel's lack of inhibition of intimal hyperplasia may be dose and/or time dependent. The inhibition of platelet aggregation and activation by clopidogrel may be of short duration, allowing for temporary inhibition for several hours, with later recurrence of platelet aggregation and activation and thus intimal hyperplasia. Higher doses may have resulted in a longer duration of inhibition, with subsequent inhibition of intimal hyperplasia. Although other studies have found that platelet aggregation and intimal hyperplasia were inhibited by clopidogrel at both preprocedural bolus doses and chronic low doses, these studies administered either larger bolus doses (10–25 mg/kg) and/or chronic doses (25 mg/kg), or they administered doses for a longer duration (16 days). 1,18 One study also revealed that larger, longer doses of clopidogrel resulted in progressively increasing inhibition of platelet aggregation and intimal hyperplasia development with a dose-response increase. 1 Harker and colleagues demonstrated that clopidogrel administered at chronic doses decreased platelet aggregation. 18 Again, the dose and duration of treatment differed from our study. Furthermore, both studies used a different animal model, which may have been associated with a less severe arterial injury.
Despite these contradictory results, multiple studies agree with our findings. Clopidogrel has been shown to decrease platelet aggregation in diabetic rats when administered as a preprocedural bolus. 17 Bolus clopidogrel also results in decreased platelet activation and aggregation after coronary stenting 8 and in patients with peripheral arterial disease. 19 Similar to our findings, Herbert and colleagues demonstrated that clopidogrel administered postprocedurally was not effective in decreasing intimal hyperplasia. 1
Although clopidogrel was effective in decreasing platelet aggregation when a preprocedural bolus dose was administered, it was not successful in inhibiting the development of intimal hyperplasia. One may speculate that factors other than platelets alone must therefore contribute to the development of intimal hyperplasia. Coller and colleagues suggested that platelet surface receptors such as glycoprotein Ib, collagen receptors, and fibrin receptors, in addition to incomplete inhibition of glycoprotein IIb/IIIa, allow for continued platelet adhesion and aggregation. 20 This suggestion is supported by clinical studies showing that 20 to 30% of patients fail to attain platelet inhibition with clopidogrel therapy 21 and that platelet activation is incompletely suppressed in patients with previous stroke treated with clopidogrel. 22 Furthermore, leukocyte activity has been implicated in the development of intimal hyperplasia. One such study suggested that leukocyte activity adjacent to arterial injury causes endothelial damage and procoagulant expression, resulting in thrombus formation. 16 Gurbel and colleagues demonstrated a persistent increase in platelet-leukocyte aggregate formation despite a preprocedural bolus of clopidogrel in patients undergoing cardiac catheterization. 8
Although the exact mechanisms are not completely elucidated by our research, clopidogrel is still a widely used, clinically useful drug. Two large randomized studies revealed that clopidogrel decreases morbid cardiovascular events in patients with preexisting cardiovascular disease. 23,24 In addition, antithrombotic drugs administered preprocedurally have been shown to improve the patency of lower extremity bypass procedures. 25 Furthermore, clopidogrel has been suggested to decrease the onset of claudicatory pain in patients with peripheral vascular disease. 26 In our study, however, with our prescribed dosing regimens, clopidogrel was not effective in decreasing intimal hyperplasia; therefore, further studies using clopidogrel are required to increase understanding of its mechanism of action and potential clinical benefits.
Footnotes
Acknowledgments
We wish to thank Mrs. Kim Henning, BS, LATG, and Mr. Wasson S. Snow, LATG, supervisor of the Veterinary Medical Unit at the Central Arkansas Veterans Healthcare System, for their invaluable assistance with this study and Mrs. Carolyn K. Wise for her assistance in determinations of homocysteine. We would also like to thank Ethicon, Inc. for supplying the suture required for this study.
Presented at the 26th World Congress of the International Society for Cardiovascular Surgery, Maui, HI, March 21–25, 2004.